
Přehled potrubí
Provozní potrubí pro hromadnou RNA-seq bylo vyvinuto jako součást řady jednotných zpracovatelských potrubí ENCODE. Úplný kód pipeline je volně k dispozici na serveru Github a lze jej spustit na serveru DNAnexus (odkaz vyžaduje vytvoření účtu) za jejich aktuální ceny.
Pipeline ENCODE Bulk RNA-seq lze použít pro replikované i nereplikované, párově zakončené i jednostranně zakončené a vláknově specifické i nevláknově specifické knihovny RNA-seq. Knihovny musí být generovány z mRNA (poly(A)+, z celkové RNA ochuzené o rRNA nebo z poly(A)-populací, které jsou velikostně selektovány tak, aby byly delší než přibližně 200 bp. V budoucnu může být tato pipeline použita také ke zpracování dat PAS-seq a Bru-seq.
Schéma pipeline pro párová data
Přehlédněte si aktuální instanci této pipeline pro párová data
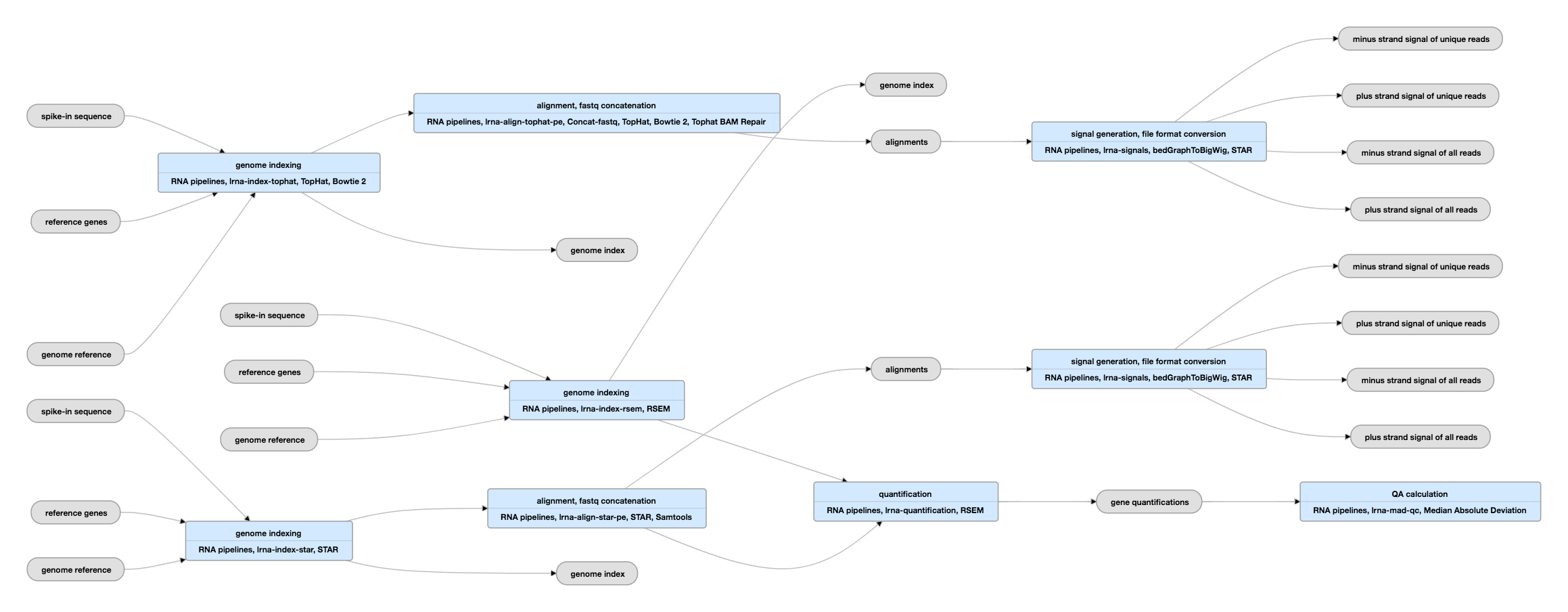
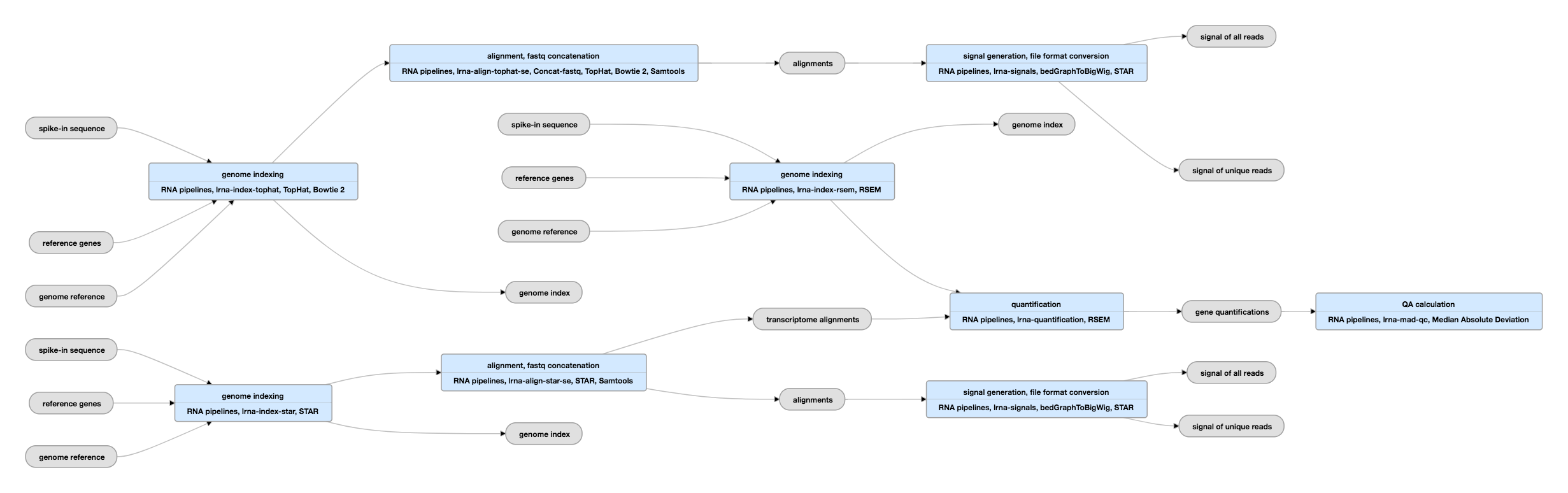
Schéma pipeline pro jednostranná data
Přehlédněte si aktuální instanci této pipeline pro jednostranná data
Vstupy:
| Formát souboru |
Informace obsažené v souboru |
Soubor popis |
Poznámky |
| fastq |
čte |
G-zazipované hromadné čtení RNA-seq | Čtení musí splňovat kritéria uvedená v části Omezení jednotné zpracovatelské linky. |
| tar | index genomu | Vytvořený programem STAR nebo TopHat | Podrobnější informace o zarovnávačích a jejich indexech naleznete v odstavci „Ohledně zarovnání a kvantifikace“ pod tabulkou „Výstupy“. |
| fasta | spike-in sekvence | ERCC Spike-ins (External RNA Control Consortium) | Spike-ins jsou ve skutečnosti kontroly pro experiment RNA-seq. |
Výstupy:
| Formát souboru |
Informace obsažené v souboru |
Popis souboru |
Poznámky |
| bam | zarovnání | Vytvořeno mapováním čtení na genom. | Podrobněji o zarovnávačích a jejich indexech viz odstavec „Pokud jde o zarovnání a kvantifikaci“ pod tabulkou „Výstupy“. |
| bam | zarovnání transkriptomu | Produkováno mapováním čtení na transkriptom. | |
| bigWig | signal | Normalizovaný signál RNA-seq | Pro vláknová data jsou generovány signály pro unikátní čtení a unikátní+multimapovací čtení v plusovém i minusovém vlákně. U nevázaných dat se signály generují pro jedinečná čtení a jedinečná+multimappingová čtení bez ohledu na identitu vlákna. |
| tsv | kvantifikace genů | Obsahuje kvantifikace hrotů |
Specifikace formátu souboru jsou následující:
|
| tsv | kvantifikace transkriptů | Obsahuje kvantifikace spike-ins | Prosím, viz upozornění týkající se kvantifikace transkriptů v odstavci níže nazvaném „Pokud jde o zarovnání a kvantifikaci“. |
| Pipeline rovněž vytváří metriky kvality, včetně Spearmanovy korelace a hloubky čtení. | |||
Ohledně zarovnání a kvantifikace:
Mapování čtení se provádí pomocí programu STAR (v některých případech se používají zarovnávače STAR i TopHat, které vytvářejí samostatné soubory bam) a kvantifikace genů a transkriptů se provádí pomocí programu RSEM. Ačkoli existuje obecná shoda mezi mapováním a kvantifikací genů provedenou různými pipeline RNA-seq, kvantifikace jednotlivých izoforem transkriptů, které jsou mnohem složitější, se mohou podstatně lišit v závislosti na použité zpracovatelské pipeline a jejich přesnost není známa. Proto lze zarovnání a kvantifikace genů používat s jistotou, zatímco kvantifikace transkriptů by se měly používat opatrně.
Genomické reference
Přehled referencí genomu a velikostí chromozomů použitých v této pipeline
Tyto pipeline vyžadují jak informace o sestavení pro druh, který je předmětem zájmu, tak genovou referenci. Každý z hlavních programů, TopHat, STAR a RSEM, vytváří index pro použití v následujících krocích. Více informací o použití programu RSEM je k dispozici zde.
Exogenní RNA spike-in kontroly
Exogenní RNA spike-in kontroly se přidávají ke vzorkům za účelem vytvoření standardní základní linie pro kvantifikaci exprese RNA (PMC3166838). Konsorcium ENCODE standardizuje používání komerčně dostupných spike-in kontrol Ambion Mix 1 v ředění ~2 % konečných mapovaných čtení. Existuje však směs starších dat a importovaných dat. Proto pro sledování spike-inů použitých v dané knihovně existuje datová sada spojená s knihovnou. Tento dataset bude obsahovat sekvenční soubor spike-inů ve formátu fasta a informace o koncentracích. Očekává se, že tyto sekvence spike-in budou nalezeny v indexu genomu použitém v kroku (krocích) mapování a v následně vygenerovaném bamu. Kvantifikace sekvencí lze nalézt v souborech kvantifikace transkriptů a genů RSEM.
Zobrazit soubory dat spike-ins
Zobrazit certifikát analýzy pro ERCC spike-ins
Přístup k nástěnce ERCC
Odkazy a publikace
Najít data vygenerovaná touto pipeline: Všechny | Pouze párové koncovky | Pouze jednokoncové koncovky
Prozkoumat publikace (probíhá)