
Oversigt over pipeline
Pipelinen Bulk RNA-seq blev udviklet som en del af ENCODE’s Uniform Processing Pipelines-serie. Den fulde pipelinekode er frit tilgængelig på Github og kan køres på DNAnexus (link kræver oprettelse af konto) til deres nuværende pris.
ENCODE Bulk RNA-seq-pipeline kan bruges til både replikerede og ureplicerede, parvis afsluttede eller enkelt afsluttede og strengspecifikke eller ikke-strengspecifikke RNA-seq-biblioteker. Biblioteker skal genereres fra mRNA (poly(A)+, rRNA-depleted total RNA eller poly(A)-populationer, der er størrelsesselekteret til at være længere end ca. 200 bp. I fremtiden kan denne pipeline også bruges til at behandle PAS-seq- og Bru-seq-data.
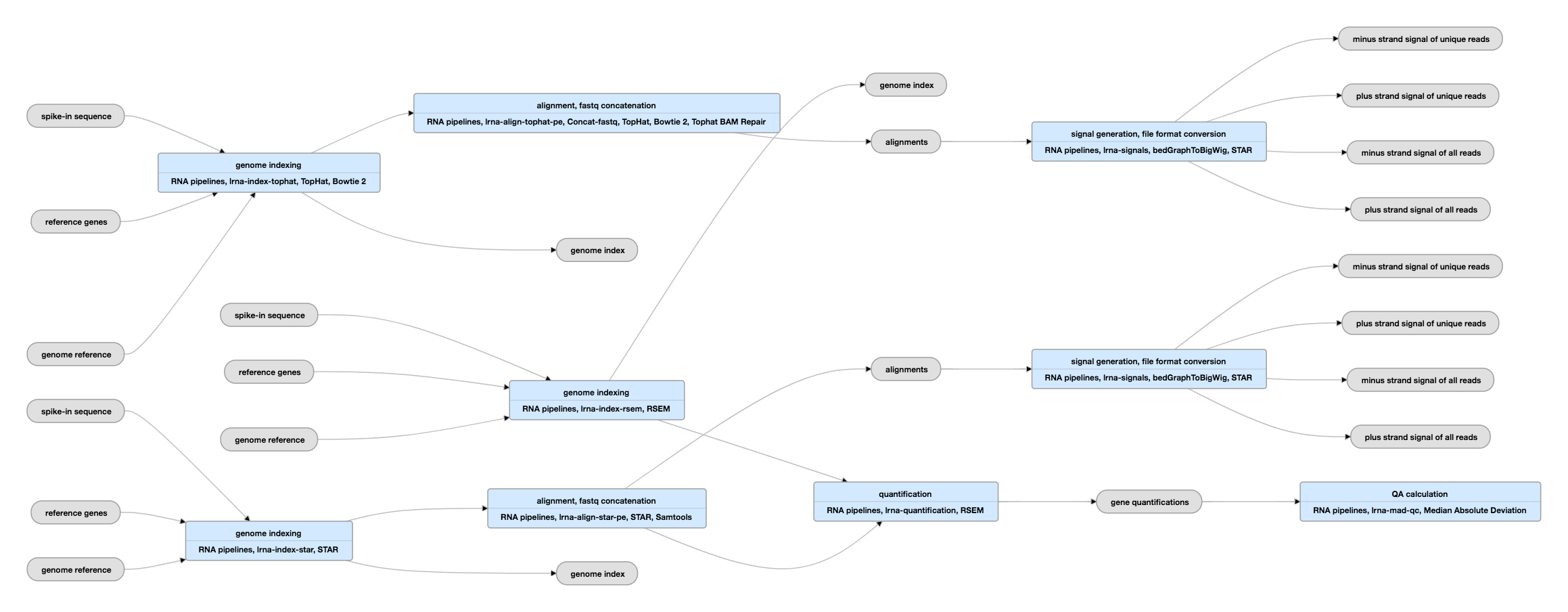
Pipeline Skematisk oversigt for parvis afsluttede data
Se den aktuelle instans af denne pipeline for parvis afsluttede data
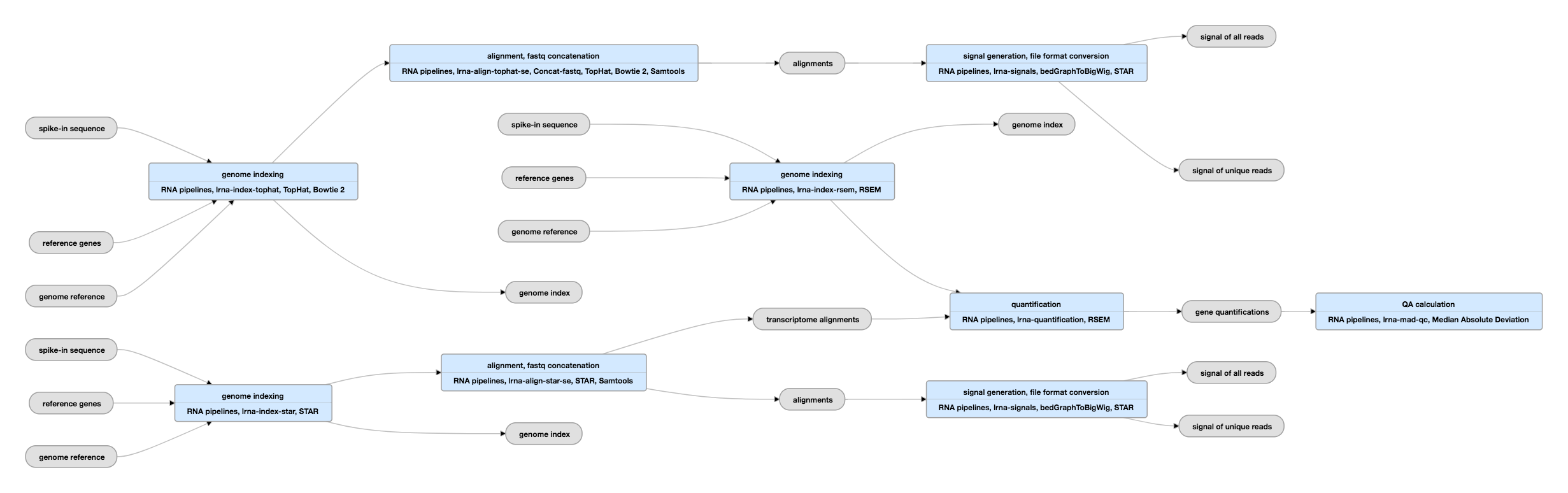
Pipeline Skematisk oversigt for single-ended data
Se de aktuelle instanser af denne pipeline for single-ended data
Input:
| Filformat |
Informationer indeholdt i filen |
Fil beskrivelse |
Noter |
|
| fastq |
læsninger |
G-zippede bulk RNA-seq-læsninger | Læsningerne skal opfylde kriterierne i begrænsningerne i den ensartede behandlingspipeline. | |
| tar | genomindeks | Genereret af STAR eller TopHat | Se afsnittet “Vedrørende alignering og kvantificering” under tabellen “Outputs” for yderligere oplysninger om alignere og deres indeks. | |
| fasta | spike-in-sekvens | ERCC Spike-ins (External RNA Control Consortium) | Spike-ins er i praksis kontrollerne for RNA-seq-eksperimentet. |
Outputs:
| Filformat |
Informationer indeholdt i filen |
Filbeskrivelse |
Notes |
| bam | alignments | Produceret ved at kortlægge læsninger til genomet. | Se venligst afsnittet “Vedrørende alignment og kvantificering” under tabellen “Outputs” for yderligere oplysninger om aligners og deres indeks. |
| bam | transcriptom alignments | Produceret ved at mappe læsninger til transkriptomet. | |
| bigWig | signal | Normaliseret RNA-seq-signal | For strandede data genereres der signaler for unikke læsninger og unikke+multimapping-læsninger i både plus- og minusstrengen. For ikke-strengede data genereres signaler for unikke læsninger og unikke+multimapping-læsninger uden hensyn til strengidentitet. |
| tsv | genkvantificeringer | Inkluderer spike-ins-kvantificeringer |
Filformatspecifikationerne er som følger:
|
| tsv | transcript quantifications | Includes the spike-ins quantifications | Se venligst advarslen vedrørende transcript quantifications i afsnittet nedenfor med titlen “Regarding alignment and quantification” (vedrørende tilpasning og kvantificering). |
| Pipelinen producerer også kvalitetsmetrikker, herunder Spearman-korrelation og læsedybde. | |||
Med hensyn til alignment og kvantificering:
Kortlægningen af læsningerne sker ved hjælp af STAR-programmet (i nogle tilfælde anvendes både STAR- og TopHat-alignere til at producere separate bam-filer), og kvantificeringen af gener og transkripter sker med RSEM-programmet. Selv om der er generel overensstemmelse mellem de kortlægninger og genkvantificeringer, der produceres af forskellige RNA-seq-pipelines, kan kvantificeringer af individuelle transkript-isoformer, der er meget mere komplekse, variere betydeligt afhængigt af den anvendte behandlingspipeline og er af ukendt nøjagtighed. Derfor kan alignments og genkvantificeringer anvendes med sikkerhed, mens transkriptkvantificeringer bør anvendes med forsigtighed.
Genomiske referencer
Se de genomreferencer og kromosomstørrelser, der anvendes i denne pipeline
Disse pipelines kræver både assemblageoplysninger for den pågældende art og en genreference. Hvert af hovedprogrammerne, TopHat, STAR og RSEM opretter et indeks til brug i de efterfølgende trin. Flere oplysninger om brugen af RSEM findes her.
Exogene RNA spike-in-kontroller
Exogene RNA spike-in-kontroller tilføjes til prøver for at skabe en standardbasislinje til kvantificering af RNA-ekspression (PMC3166838). ENCODE-konsortiet standardiserer brugen af Ambion Mix 1 kommercielt tilgængelige spike-ins i en fortynding på ~2 % af de endelige kortlagte læsninger. Der er dog tale om en blanding af ældre data og importerede data. For at spore de spike-ins, der er anvendt i et givet bibliotek, er der derfor et datasæt knyttet til biblioteket. Dette datasæt vil indeholde spike-ins-sekvensfilen i fasta-format og oplysninger om koncentrationerne. Disse spike-ins-sekvenser forventes at blive fundet i det genomindeks, der anvendes i kortlægningsfasen(erne), og i den efterfølgende genererede bam-fil. Kvantificeringerne af sekvenserne kan findes i RSEM-transkript- og genkvantificeringsfilerne.
Se spike-ins-datasæt
Se analysecertifikatet for ERCC spike-ins
Access the ERCC dash board
Links and Publications
Søg data genereret af denne pipeline: Alle | kun parrede ender | kun en ende
Udforsk publikationer (under udarbejdelse)