
- Genom Sequencing
- Genetisk karakterisering
- Metoder til sekventering af influenzavirusgenomet
Genome Sequencing
Influenzavirus ændrer sig konstant, faktisk undergår alle influenzavirus genetiske ændringer over tid (for yderligere oplysninger, se Hvordan influenzavirus kan ændre sig: “Drift” og “Shift”). Et influenzavirus’ genom består af alle de gener, der udgør viruset. CDC fører året rundt overvågning af cirkulerende influenzavirus for at overvåge ændringer i genomet (eller dele af genomet) af disse vira. Dette arbejde udføres som en del af den rutinemæssige amerikanske influenzaovervågning og som en del af CDC’s rolle som et af Verdenssundhedsorganisationens (WHO) samarbejdscentre for reference og forskning om influenza. De oplysninger, som CDC indsamler ved at studere genetiske ændringer (også kendt som “substitutioner”, “varianter” eller “mutationer”) i influenzavirus, spiller en vigtig rolle for folkesundheden ved at hjælpe med at afgøre, om vacciner og antivirale lægemidler vil virke mod de aktuelt cirkulerende influenzavirus, og ved at hjælpe med at afgøre, om influenzavirus hos dyr kan inficere mennesker.
Genomsekventering afslører sekvensen af nukleotiderne i et gen, ligesom alfabetets bogstaver i ord. Nukleotider er organiske molekyler, der udgør den strukturelle byggesten i nukleinsyrer, f.eks. RNA eller DNA. Alle influenzavirus består af enkeltstrenget RNA i modsætning til dobbeltstrenget DNA. Influenzavirus’ RNA-gener består af kæder af nukleotider, der er bundet sammen og kodet af bogstaverne A, C, G og U, som står for henholdsvis adenin, cytosin, guanin og uracil. Ved at sammenligne sammensætningen af nukleotider i et virusgen med rækkefølgen af nukleotider i et andet virusgen kan man afsløre variationer mellem de to vira.
Genetiske variationer er vigtige, fordi de kan påvirke strukturen af en influenzavirus’ overfladeproteiner. Proteiner består af sekvenser af aminosyrer.
Substitutionen af en aminosyre med en anden kan påvirke egenskaberne ved en virus, f.eks. hvor godt en virus overføres mellem mennesker, og hvor modtagelig virussen er over for antivirale lægemidler eller nuværende vacciner.
![]()
![]()
Genomsekventering afslører rækkefølgen af nukleotiderne i et gen, ligesom alfabetets bogstaver i ord. Ved at sammenligne sammensætningen af nukleotider i et virusgen med rækkefølgen af nukleotider i et andet virusgen kan man afsløre variationer mellem de to vira.
Genetiske variationer er vigtige, fordi de påvirker strukturen af en influenzavirus’ overfladeproteiner. Proteiner består af sekvenser af aminosyrer.
Substitutionen af en aminosyre med en anden kan påvirke en viruss egenskaber, f.eks. hvor godt en virus overføres mellem mennesker, og hvor modtagelig virussen er over for antivirale lægemidler eller de nuværende vacciner.
Influenza A- og B-virus – de primære influenzavirus, der inficerer mennesker – er RNA-virus, der har otte gen-segmenter. Disse gener indeholder “instruktioner” til fremstilling af nye vira, og det er disse instruktioner, som en influenzavirus bruger, når den inficerer en menneskelig celle, til at narre cellen til at producere flere influenzavira og derved sprede infektionen.
Influenzagenerne består af en sekvens af molekyler kaldet nukleotider, der binder sig sammen i en kædeform. Nukleotider betegnes med bogstaverne A, C, G og U.
Genomsekventering er en proces, der bestemmer rækkefølgen, eller sekvensen, af nukleotiderne (dvs. A, C, G og U) i hvert af de gener, der findes i virusets genom. Ved sekventering af hele genomet kan man afsløre den ca. 13 500 bogstaver store sekvens af alle generne i virusets genom.
Hvert år udfører CDC sekventering af hele genomet på ca. 7 000 influenzavirus fra originale kliniske prøver, der er indsamlet i forbindelse med virologisk overvågning. Et influenza A- eller B-virus’ genom indeholder otte gen-segmenter, der koder for (dvs. bestemmer strukturen og egenskaberne ved) virusets 12 proteiner, herunder de to primære overfladeproteiner: hæmagglutinin (HA) og neuraminidase (NA). En influenzavirus’ overfladeproteiner bestemmer vigtige egenskaber ved viruset, herunder hvordan viruset reagerer på visse antivirale lægemidler, virusets genetiske lighed med de nuværende influenzavaccinevirus og muligheden for, at zoonotiske (af animalsk oprindelse) influenzavirus kan inficere menneskelige værter.
Genetisk karakterisering
CDC og andre folkesundhedslaboratorier rundt om i verden har sekventeret generne fra influenzavirus siden 1980’erne. CDC bidrager med gensekvenser til offentlige databaser, f.eks. GenBankexternt ikon og Global Initiative on Sharing Avian Influenza Data (GISAID)eksternt ikon, til brug for forskere inden for folkesundhed. De resulterende biblioteker af gensekvenser gør det muligt for CDC og andre laboratorier at sammenligne generne i de aktuelt cirkulerende influenzavirus med generne i ældre influenzavirus og i de virus, der anvendes i vacciner. Denne proces med sammenligning af genetiske sekvenser kaldes genetisk karakterisering. CDC bruger genetisk karakterisering af følgende grunde:
- For at bestemme, hvor tæt “beslægtede” eller ens influenzavirusene er med hinanden genetisk
- For at overvåge, hvordan influenzavirusene udvikler sig
- For at identificere genetiske ændringer, der påvirker virusets egenskaber. F.eks. at identificere de specifikke ændringer, der er forbundet med influenzavirus, som spreder sig lettere, forårsager mere alvorlig sygdom eller udvikler resistens over for antivirale lægemidler
- At vurdere, hvor godt en influenzavaccine kan beskytte mod et bestemt influenzavirus på grundlag af dets genetiske lighed med viruset
- At overvåge, om der er genetiske ændringer i influenzavirus, der cirkulerer i dyrepopulationer, som kan gøre dem i stand til at inficere mennesker.
De relative forskelle mellem en gruppe influenzavirus vises ved at organisere dem i en grafik kaldet et “fylogenetisk træ”. Fylogenetiske træer for influenzavirus er ligesom stamtræer (slægtsforskning) for mennesker. Disse træer viser, hvor tæt “beslægtede” de enkelte vira er med hinanden. Virusene grupperes sammen på grundlag af, om deres geners nukleotider er identiske eller ej. Fylogenetiske træer for influenzavirus viser normalt, hvor ens virusenes hæmagglutinin- (HA) eller neuraminidase- (NA) gener er med hinanden. Hver sekvens fra en specifik influenzavirus har sin egen gren på træet. Graden af genetisk forskel (antallet af nukleotidforskelle) mellem virusene repræsenteres af længden af de vandrette linjer (grene) i det fylogenetiske træ. Jo længere virusene ligger fra hinanden på den vandrette akse i et fylogenetisk træ, jo mere genetisk forskellige er virusene fra hinanden.
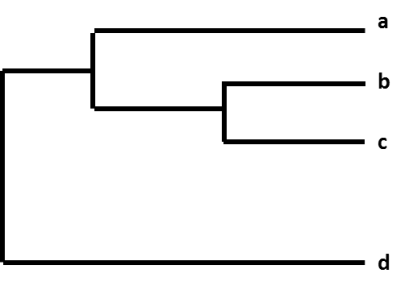
Figur. Et fylogenetisk træ.
For eksempel, efter at CDC har sekventeret en influenza A(H3N2)-virus, der er indsamlet ved overvågning, katalogiseres virussekvensen sammen med andre virussekvenser, der har et lignende HA-gen (H3) og et lignende NA-gen (N2). Som led i denne proces sammenligner CDC den nye virussekvens med de andre virussekvenser og undersøger, om der er forskelle mellem dem. CDC bruger derefter et fylogenetisk træ til visuelt at repræsentere, hvor genetisk forskellige A(H3N2)-virusserne er fra hinanden.
CDC udfører genetisk karakterisering af influenzavirus året rundt. Disse genetiske data bruges sammen med data om antigenisk karakterisering af virus til at hjælpe med at afgøre, hvilke vaccinevirus der skal vælges til de kommende influenzavacciner på den nordlige eller sydlige halvkugle. I månederne op til WHO’s vaccinkonsultationsmøder i februar og september indsamler CDC influenzavirus gennem overvågning og sammenligner HA- og NA-gensekvenserne i de nuværende vaccinevirus med sekvenserne i cirkulerende influenzavirus. Dette er en måde at vurdere, hvor tæt beslægtet de cirkulerende influenzavirus er med de vira, som den sæsonbestemte influenzavaccine blev udviklet til at beskytte mod. Efterhånden som virus indsamles og karakteriseres genetisk, kan forskelle afsløres.
For eksempel vil cirkulerende virus undertiden i løbet af en sæson ændre sig genetisk, hvilket gør, at de adskiller sig fra det tilsvarende vaccinevirus. Dette er et tegn på, at det kan være nødvendigt at vælge et andet vaccinevirus til næste influenzasæsons vaccine, selv om andre faktorer, herunder resultaterne af den antigeniske karakterisering, i høj grad påvirker vaccinebeslutningerne. HA- og NA-overfladeproteinerne i influenzavirus er antigener, hvilket betyder, at de genkendes af immunsystemet og er i stand til at udløse et immunrespons, herunder produktion af antistoffer, der kan blokere infektion. Antigenisk karakterisering henviser til analysen af en viruss reaktion med antistoffer for at hjælpe med at vurdere, hvordan den forholder sig til en anden virus.
Metoder til sekventering af influenzavirusgenomet
En influenzaprøve indeholder mange influenzaviruspartikler, der blev dyrket i et reagensglas, og som ofte har små genetiske forskelle i forhold til hinanden blandt hele populationen af søskendevirus.
Traditionelt har forskerne brugt en sekventeringsteknik kaldet “Sanger-reaktionen” til at overvåge influenzaudviklingen som en del af den virologiske overvågning. Sanger-sekventering identificerer den fremherskende genetiske sekvens blandt de mange influenzavirus, der findes i et isolat. Det betyder, at små variationer i populationen af virusser i en prøve ikke afspejles i det endelige resultat. Forskere bruger ofte Sanger-metoden til at foretage delvis genomsekventering af influenzavirus, mens nyere teknologier (se næste afsnit) er bedre egnet til sekventering af hele genomet.
I løbet af de sidste fem år har CDC anvendt “Next Generation Sequencing (NGS)-metoder”, som i høj grad har udvidet mængden af oplysninger og detaljer, som sekventeringsanalyser kan give. NGS anvender avanceret molekylær detektion (AMD) til at identificere gensekvenser fra hver enkelt virus i en prøve. Derfor afslører NGS de genetiske variationer blandt mange forskellige influenzaviruspartikler i en enkelt prøve, og disse metoder afslører også hele den kodende region i genomerne. Denne detaljeringsgrad kan direkte gavne beslutningstagningen på folkesundhedsområdet på vigtige måder, men dataene skal fortolkes omhyggeligt af højtuddannede eksperter i sammenhæng med andre tilgængelige oplysninger. Se AMD-projekter: Forbedring af influenzavacciner for at få flere oplysninger om, hvordan NGS og AMD er ved at revolutionere kortlægningen af influenzagenomet hos CDC.