
Pipeline Übersicht
Die Bulk RNA-seq Pipeline wurde als Teil der ENCODE Uniform Processing Pipelines Serie entwickelt. Der vollständige Pipeline-Code ist auf Github frei verfügbar und kann auf DNAnexus (Link erfordert die Erstellung eines Kontos) zu deren aktuellen Preisen ausgeführt werden.
Die ENCODE Bulk RNA-seq Pipeline kann sowohl für replizierte als auch für nicht replizierte, paired-ended oder single-ended und strangspezifische oder nicht strangspezifische RNA-seq Bibliotheken verwendet werden. Die Bibliotheken müssen aus mRNA (poly(A)+, rRNA-verarmter Gesamt-RNA oder poly(A)-Populationen generiert werden, die so ausgewählt werden, dass sie länger als etwa 200 bp sind. In Zukunft kann diese Pipeline auch für die Verarbeitung von PAS-seq- und Bru-seq-Daten verwendet werden.
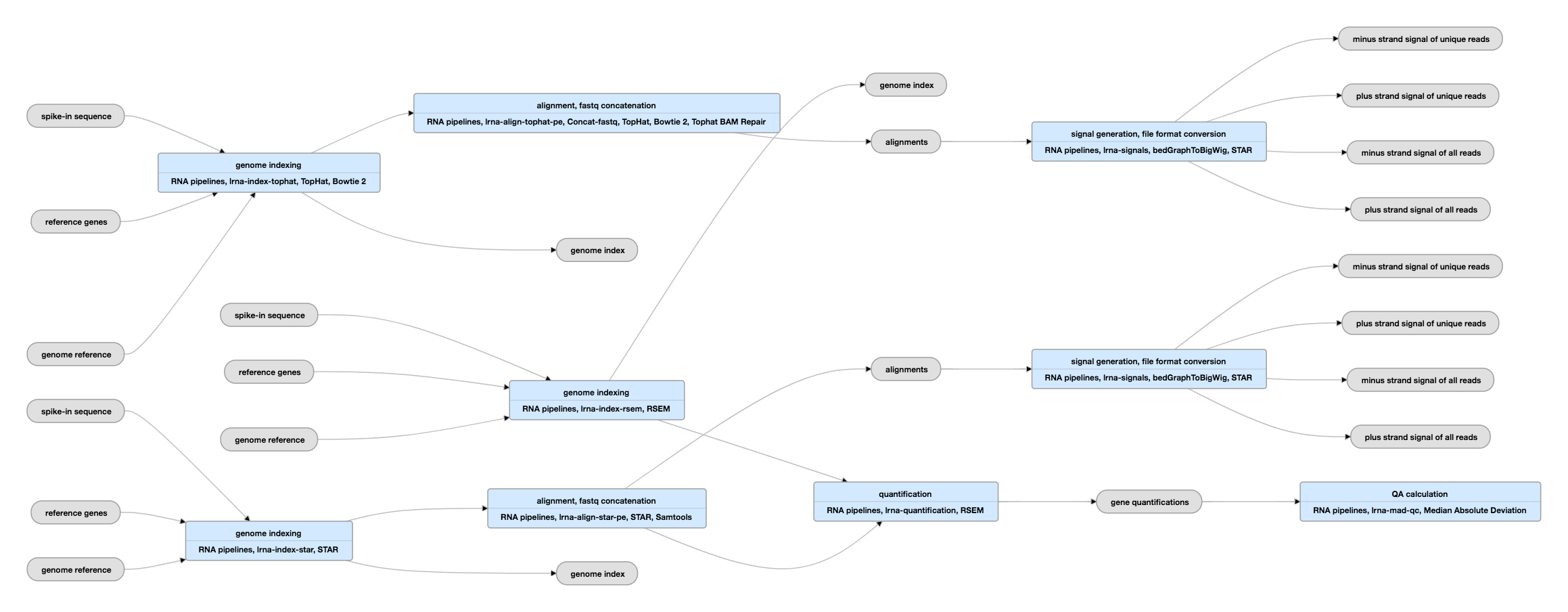
Pipeline-Schema für paired-ended Daten
Ansicht der aktuellen Instanz dieser Pipeline für paired-ended Daten
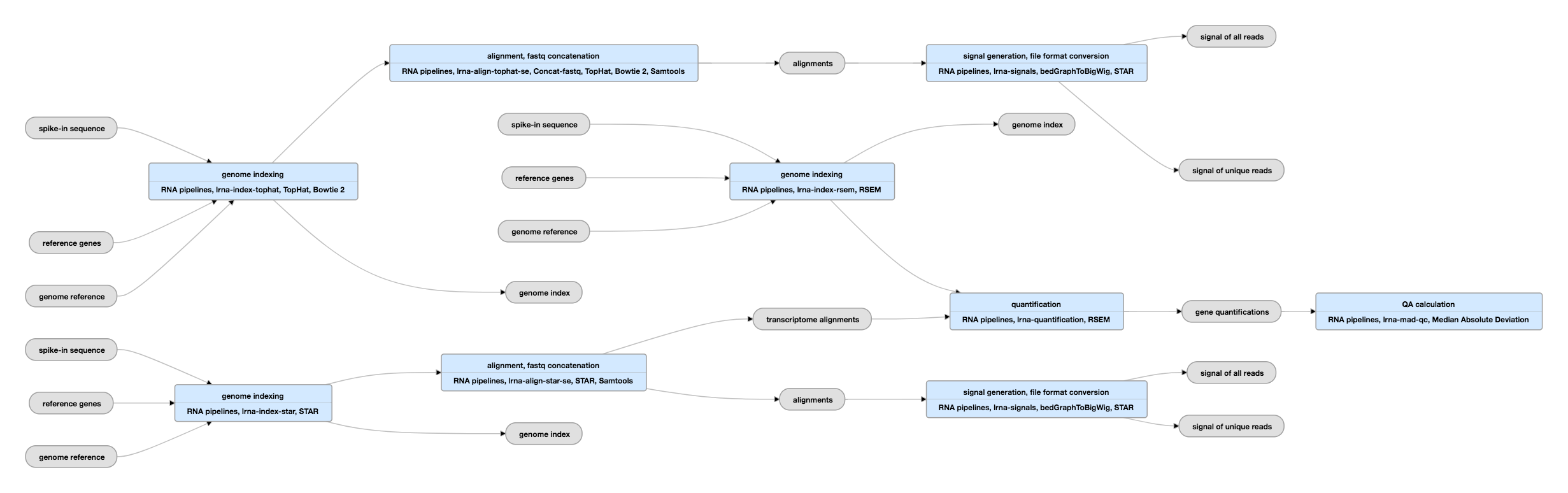
Pipeline-Schema für single-ended Daten
Ansicht der aktuellen Instanzen dieser Pipeline für single-ended Daten
Eingaben:
| Dateiformat |
Informationen in Datei enthalten |
Datei Beschreibung |
Hinweise |
| fastq |
Lesungen |
G-gezippte Bulk-RNA-seq-Reads | Die Reads müssen die in den „Uniform Processing Pipeline Restrictions“ genannten Kriterien erfüllen. |
| tar | Genom-Index | Generiert von STAR oder TopHat | Weitere Informationen zu den Alignern und ihren Indizes finden Sie im Abschnitt „Betreffend Alignment und Quantifizierung“ unter der Tabelle „Outputs“. |
| fasta | Spike-in-Sequenz | ERCC Spike-ins (External RNA Control Consortium) | Die Spike-ins sind quasi die Kontrollen für das RNA-seq-Experiment. |
Outputs:
| Dateiformat |
Informationen in der Datei enthalten |
Dateibeschreibung |
Hinweise |
| bam | Ausrichtungen | Erzeugt durch Mapping von Reads auf das Genom. | Weitere Informationen zu den Alignern und ihren Indizes finden Sie im Abschnitt „Zum Alignment und zur Quantifizierung“ unter der Tabelle „Outputs“. |
| bam | Transkriptom-Alignments | Erzeugt durch Mapping von Reads auf das Transkriptom. | |
| bigWig | signal | Normalisiertes RNA-seq-Signal | Für gestrandete Daten werden Signale für Unique Reads und Unique+Multimapping Reads im Plus- und Minus-Strang erzeugt. Bei nicht-strängigen Daten werden Signale für eindeutige Reads und eindeutige+Multimapping-Reads ohne Berücksichtigung der Strangidentität erzeugt. |
| tsv | Gen-Quantifizierungen | Enthält die Spike-Ins-Quantifizierungen |
Das Dateiformat ist wie folgt spezifiziert:
|
| tsv | Transkript-Quantifizierungen | Einschließlich der Spike-Ins-Quantifizierungen | Bitte beachten Sie den Hinweis zu den Transkript-Quantifizierungen im Abschnitt „Zum Alignment und zur Quantifizierung“ unten. |
| Die Pipeline erzeugt auch Qualitätsmetriken, einschließlich Spearman-Korrelation und Lesetiefe. | |||
Zum Alignment und zur Quantifizierung:
Das Mapping der Reads erfolgt mit dem STAR-Programm (in einigen Fällen werden sowohl STAR- als auch TopHat-Aligner verwendet, um separate bam-Dateien zu erzeugen) und die Quantifizierung von Genen und Transkripten erfolgt mit dem RSEM-Programm. Obwohl es eine allgemeine Übereinstimmung zwischen den Mappings und den Genquantifizierungen gibt, die von verschiedenen RNA-seq-Pipelines erzeugt werden, können die Quantifizierungen einzelner Transkript-Isoformen, die viel komplexer sind, je nach verwendeter Verarbeitungspipeline erheblich voneinander abweichen und sind von unbekannter Genauigkeit. Daher können Alignments und Gen-Quantifizierungen sicher verwendet werden, während Transkript-Quantifizierungen mit Vorsicht verwendet werden sollten.
Genomische Referenzen
Betrachten Sie die Genom-Referenzen und Chromosomengrößen, die in dieser Pipeline verwendet werden
Diese Pipelines erfordern sowohl Assembler-Informationen für die interessierende Art als auch eine Gen-Referenz. Jedes der Hauptprogramme, TopHat, STAR und RSEM, erstellt einen Index, der in den nachfolgenden Schritten verwendet wird. Weitere Informationen über die Verwendung von RSEM finden Sie hier.
Exogene RNA-Spike-In-Kontrollen
Exogene RNA-Spike-In-Kontrollen werden zu den Proben hinzugefügt, um eine Standard-Basislinie für die Quantifizierung der RNA-Expression zu schaffen (PMC3166838). Das ENCODE-Konsortium verwendet standardmäßig die im Handel erhältlichen Ambion Mix 1 Spike-Ins in einer Verdünnung von ~2 % der endgültigen kartierten Reads. Es gibt jedoch eine Mischung aus älteren Daten und importierten Daten. Um die in einer bestimmten Bibliothek verwendeten Spike-Ins zu verfolgen, gibt es daher einen mit der Bibliothek verbundenen Datensatz. Dieser Datensatz enthält die Spike-In-Sequenzdatei im Fasta-Format und Informationen zu den Konzentrationen. Es wird erwartet, dass diese Spike-In-Sequenzen im Genom-Index, der in den Mapping-Schritten verwendet wurde, und in der anschließend generierten bam-Datei zu finden sind. Die Quantifizierungen der Sequenzen sind in den RSEM-Transkript- und Genquantifizierungsdateien zu finden.
Spike-Ins-Datensätze anzeigen
Analysezertifikat für ERCC-Spike-Ins anzeigen
Zugriff auf das ERCC-Dashboard
Links und Veröffentlichungen
Mit dieser Pipeline erzeugte Daten finden: Alle | Nur Paired-End | Nur Single-End
Erkunden Sie Veröffentlichungen (in Arbeit)