
- Genomsequenzierung Sequenzierung
- Genetische Charakterisierung
- Methoden der Grippe-Genomsequenzierung
Genomsequenzierung
Influenzaviren verändern sich ständig, Tatsächlich unterliegen alle Influenzaviren im Laufe der Zeit genetischen Veränderungen (weitere Informationen finden Sie unter Wie sich das Grippevirus verändern kann: „Drift“ und „Shift“). Das Genom eines Influenzavirus besteht aus allen Genen, aus denen das Virus besteht. Das CDC überwacht ganzjährig zirkulierende Influenzaviren, um Veränderungen des Genoms (oder von Teilen des Genoms) dieser Viren zu beobachten. Diese Arbeit erfolgt im Rahmen der routinemäßigen Influenza-Überwachung in den USA und als Teil der Rolle des CDC als Kooperationszentrum der Weltgesundheitsorganisation (WHO) für Referenz und Influenzaforschung. Die Informationen, die das CDC durch die Untersuchung genetischer Veränderungen (auch bekannt als „Substitutionen“, „Varianten“ oder „Mutationen“) in Influenzaviren sammelt, spielen eine wichtige Rolle für die öffentliche Gesundheit, da sie dazu beitragen, festzustellen, ob Impfstoffe und antivirale Medikamente gegen die derzeit zirkulierenden Influenzaviren wirken, und helfen, das Potenzial von Influenzaviren in Tieren zu bestimmen, Menschen zu infizieren.
Die Sequenzierung des Genoms offenbart die Abfolge der Nukleotide in einem Gen, wie die Buchstaben in einem Wort. Nukleotide sind organische Moleküle, die den strukturellen Grundbaustein von Nukleinsäuren, wie RNA oder DNA, bilden. Alle Grippeviren bestehen aus einzelsträngiger RNA im Gegensatz zu doppelsträngiger DNA. Die RNA-Gene der Influenzaviren bestehen aus Ketten von Nukleotiden, die miteinander verbunden sind und durch die Buchstaben A, C, G und U kodiert werden, die jeweils für Adenin, Cytosin, Guanin und Uracil stehen. Vergleicht man die Zusammensetzung der Nukleotide in einem Virusgen mit der Reihenfolge der Nukleotide in einem anderen Virusgen, kann man Variationen zwischen den beiden Viren feststellen.
Genetische Variationen sind wichtig, weil sie die Struktur der Oberflächenproteine eines Influenzavirus beeinflussen können. Proteine bestehen aus Sequenzen von Aminosäuren.
Die Substitution einer Aminosäure durch eine andere kann die Eigenschaften eines Virus beeinflussen, z. B. wie gut ein Virus zwischen Menschen übertragen werden kann und wie anfällig das Virus für antivirale Medikamente oder aktuelle Impfstoffe ist.
![]()
![]()
Genomsequenzierung offenbart die Abfolge der Nukleotide in einem Gen, wie die Buchstaben in einem Wort. Der Vergleich der Zusammensetzung der Nukleotide in einem Virusgen mit der Reihenfolge der Nukleotide in einem anderen Virusgen kann Variationen zwischen den beiden Viren aufdecken.
Genetische Variationen sind wichtig, weil sie die Struktur der Oberflächenproteine eines Influenzavirus beeinflussen. Proteine bestehen aus Aminosäuresequenzen.
Der Austausch einer Aminosäure gegen eine andere kann die Eigenschaften eines Virus beeinflussen, z. B. wie gut ein Virus zwischen Menschen übertragen werden kann und wie anfällig das Virus für antivirale Medikamente oder aktuelle Impfstoffe ist.
Influenza A- und B-Viren – die primären Influenzaviren, die Menschen infizieren – sind RNA-Viren, die acht Gensegmente haben. Diese Gene enthalten „Anweisungen“ für die Herstellung neuer Viren, und es sind diese Anweisungen, die ein Influenzavirus verwendet, sobald es eine menschliche Zelle infiziert hat, um die Zelle dazu zu bringen, mehr Influenzaviren zu produzieren und so die Infektion zu verbreiten.
Influenzagene bestehen aus einer Abfolge von Molekülen, die als Nukleotide bezeichnet werden und in einer kettenartigen Form aneinander gebunden sind. Die Nukleotide werden mit den Buchstaben A, C, G und U bezeichnet.
Bei der Genomsequenzierung wird die Reihenfolge oder Sequenz der Nukleotide (d. h. A, C, G und U) in jedem der Gene im Genom des Virus bestimmt. Die vollständige Genomsequenzierung kann die aus etwa 13.500 Buchstaben bestehende Sequenz aller Gene des Virusgenoms aufdecken.
Jedes Jahr führt das CDC die vollständige Genomsequenzierung an etwa 7.000 Influenzaviren aus klinischen Originalproben durch, die im Rahmen der virologischen Überwachung gesammelt wurden. Das Genom eines Influenza-A- oder -B-Virus enthält acht Gensegmente, die für die 12 Proteine des Virus kodieren (d. h. deren Struktur und Eigenschaften bestimmen), darunter die beiden wichtigsten Oberflächenproteine: Hämagglutinin (HA) und Neuraminidase (NA). Die Oberflächenproteine eines Influenzavirus bestimmen wichtige Eigenschaften des Virus, wie z. B. die Reaktion des Virus auf bestimmte antivirale Medikamente, die genetische Ähnlichkeit des Virus mit aktuellen Influenza-Impfviren und das Potenzial von zoonotischen (von Tieren stammenden) Influenzaviren, menschliche Wirte zu infizieren.
Genetische Charakterisierung
Das CDC und andere Laboratorien des öffentlichen Gesundheitswesens auf der ganzen Welt sequenzieren seit den 1980er Jahren die Gene von Influenzaviren. Das CDC stellt Gensequenzen für öffentliche Datenbanken wie GenBankexternes Symbol und die Global Initiative on Sharing Avian Influenza Data (GISAID)externes Symbol zur Verfügung, die von Forschern des öffentlichen Gesundheitswesens genutzt werden können. Die daraus resultierenden Gensequenzbibliotheken ermöglichen es dem CDC und anderen Labors, die Gene der derzeit zirkulierenden Influenzaviren mit den Genen älterer Influenzaviren und den in Impfstoffen verwendeten Viren zu vergleichen. Dieser Prozess des Vergleichs von Gensequenzen wird als genetische Charakterisierung bezeichnet. Das CDC verwendet die genetische Charakterisierung aus folgenden Gründen:
- Um festzustellen, wie eng „verwandt“ oder ähnlich Grippeviren genetisch miteinander sind
- Um zu beobachten, wie sich Grippeviren weiterentwickeln
- Um genetische Veränderungen zu identifizieren, die die Eigenschaften des Virus beeinflussen. Zum Beispiel, um die spezifischen Veränderungen zu identifizieren, die damit verbunden sind, dass sich Influenzaviren leichter ausbreiten, schwerere Krankheiten verursachen oder eine Resistenz gegen antivirale Medikamente entwickeln
- Um zu beurteilen, wie gut ein Grippeimpfstoff gegen ein bestimmtes Influenzavirus auf der Grundlage seiner genetischen Ähnlichkeit mit dem Virus schützen könnte
- Um nach genetischen Veränderungen bei Influenzaviren zu suchen, die in Tierpopulationen zirkulieren und es ihnen ermöglichen könnten, Menschen zu infizieren.
Die relativen Unterschiede zwischen einer Gruppe von Influenzaviren werden in einer Grafik dargestellt, die als „phylogenetischer Baum“ bezeichnet wird. Phylogenetische Bäume für Influenzaviren sind wie Stammbäume (Genealogie) für Menschen. Diese Bäume zeigen, wie eng die einzelnen Viren miteinander „verwandt“ sind. Die Viren werden danach gruppiert, ob die Nukleotide ihrer Gene identisch sind oder nicht. Phylogenetische Bäume von Influenzaviren zeigen in der Regel an, wie ähnlich die Hämagglutinin (HA)- oder Neuraminidase (NA)-Gene der Viren einander sind. Jede Sequenz eines bestimmten Influenzavirus hat ihren eigenen Ast im Baum. Der Grad der genetischen Unterschiede (Anzahl der Nukleotidunterschiede) zwischen den Viren wird durch die Länge der horizontalen Linien (Äste) im phylogenetischen Baum dargestellt. Je weiter die Viren auf der horizontalen Achse eines phylogenetischen Baums voneinander entfernt sind, desto stärker unterscheiden sie sich genetisch voneinander.
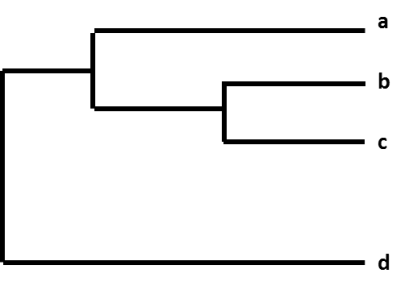
Abbildung. Ein phylogenetischer Baum.
Nach der Sequenzierung eines Influenza-A(H3N2)-Virus durch das CDC, das im Rahmen der Überwachung gesammelt wurde, wird die Virussequenz mit anderen Virussequenzen katalogisiert, die ein ähnliches HA-Gen (H3) und ein ähnliches NA-Gen (N2) aufweisen. Im Rahmen dieses Prozesses vergleicht die CDC die neue Virensequenz mit den anderen Virensequenzen und sucht nach Unterschieden zwischen ihnen. CDC verwendet dann einen phylogenetischen Baum, um visuell darzustellen, wie sehr sich die A(H3N2)-Viren genetisch voneinander unterscheiden.
CDC führt das ganze Jahr über eine genetische Charakterisierung von Grippeviren durch. Diese genetischen Daten werden in Verbindung mit den Daten zur Antigencharakterisierung der Viren verwendet, um zu bestimmen, welche Impfviren für die kommenden Grippeimpfstoffe der nördlichen und südlichen Hemisphäre ausgewählt werden sollten. In den Monaten vor den WHO-Konsultationssitzungen zu Impfstoffen im Februar und September sammelt das CDC Influenzaviren durch Überwachung und vergleicht die HA- und NA-Gensequenzen aktueller Impfviren mit denen zirkulierender Grippeviren. Auf diese Weise lässt sich beurteilen, wie eng die zirkulierenden Grippeviren mit den Viren verwandt sind, gegen die der saisonale Grippeimpfstoff entwickelt wurde. Wenn Viren gesammelt und genetisch charakterisiert werden, können Unterschiede aufgedeckt werden.
So verändern sich z. B. zirkulierende Viren manchmal im Laufe einer Saison genetisch, so dass sie sich von dem entsprechenden Impfvirus unterscheiden. Dies ist ein Hinweis darauf, dass für den Impfstoff der nächsten Grippesaison möglicherweise ein anderes Impfvirus ausgewählt werden muss, obwohl andere Faktoren, einschließlich der Ergebnisse der Antigencharakterisierung, die Impfstoffentscheidungen stark beeinflussen. Die HA- und NA-Oberflächenproteine von Influenzaviren sind Antigene, d. h. sie werden vom Immunsystem erkannt und können eine Immunreaktion auslösen, einschließlich der Produktion von Antikörpern, die eine Infektion verhindern können. Die antigene Charakterisierung bezieht sich auf die Analyse der Reaktion eines Virus mit Antikörpern, um zu beurteilen, wie es mit einem anderen Virus verwandt ist.
Methoden der Grippe-Genom-Sequenzierung
Eine Influenza-Probe enthält viele Influenza-Viruspartikel, die in einem Reagenzglas gezüchtet wurden und die oft nur geringe genetische Unterschiede im Vergleich zu der gesamten Population der Geschwisterviren aufweisen.
Traditionell haben Wissenschaftler eine Sequenzierungstechnik namens „Sanger-Reaktion“ verwendet, um die Influenza-Evolution als Teil der virologischen Überwachung zu verfolgen. Die Sanger-Sequenzierung identifiziert die vorherrschende genetische Sequenz unter den vielen in einem Isolat gefundenen Influenzaviren. Das bedeutet, dass kleine Variationen in der Population der in einer Probe vorhandenen Viren im Endergebnis nicht berücksichtigt werden. Wissenschaftler verwenden die Sanger-Methode häufig, um eine partielle Genomsequenzierung von Influenzaviren durchzuführen, während neuere Technologien (siehe nächster Absatz) besser für die Sequenzierung des gesamten Genoms geeignet sind.
In den letzten fünf Jahren hat das CDC „Next Generation Sequencing (NGS)“-Methoden eingesetzt, die die Menge an Informationen und Details, die die Sequenzierungsanalyse liefern kann, erheblich erweitert haben. NGS verwendet eine fortschrittliche molekulare Detektion (AMD), um Gensequenzen von jedem Virus in einer Probe zu identifizieren. NGS enthüllt daher die genetischen Variationen zwischen vielen verschiedenen Influenzaviruspartikeln in einer einzigen Probe, und diese Methoden enthüllen auch die gesamte kodierende Region der Genome. Diese Detailgenauigkeit kann für die Entscheidungsfindung im Bereich der öffentlichen Gesundheit von großem Nutzen sein, aber die Daten müssen von hochqualifizierten Experten im Kontext anderer verfügbarer Informationen sorgfältig interpretiert werden. Siehe AMD-Projekte: Verbesserung von Grippeimpfstoffen für weitere Informationen darüber, wie NGS und AMD die Kartierung des Grippegenoms bei der CDC revolutionieren.