
Resumen de la tubería
La tubería de ARN-seq a granel fue desarrollada como parte de la serie de tuberías de procesamiento uniforme de ENCODE. El código completo del pipeline está disponible de forma gratuita en Github y se puede ejecutar en DNAnexus (el enlace requiere la creación de una cuenta) a su precio actual.
La tubería ENCODE Bulk RNA-seq puede ser utilizada para bibliotecas de RNA-seq replicadas y no replicadas, emparejadas o de un solo extremo, y de cadenas específicas o no específicas. Las bibliotecas deben generarse a partir de poblaciones de ARNm (poli(A)+, ARN total empobrecido en ARNr, o poli(A)- que se seleccionan por su tamaño para que sean más largas que aproximadamente 200 pb. En el futuro, esta tubería también puede utilizarse para procesar datos de PAS-seq y Bru-seq.
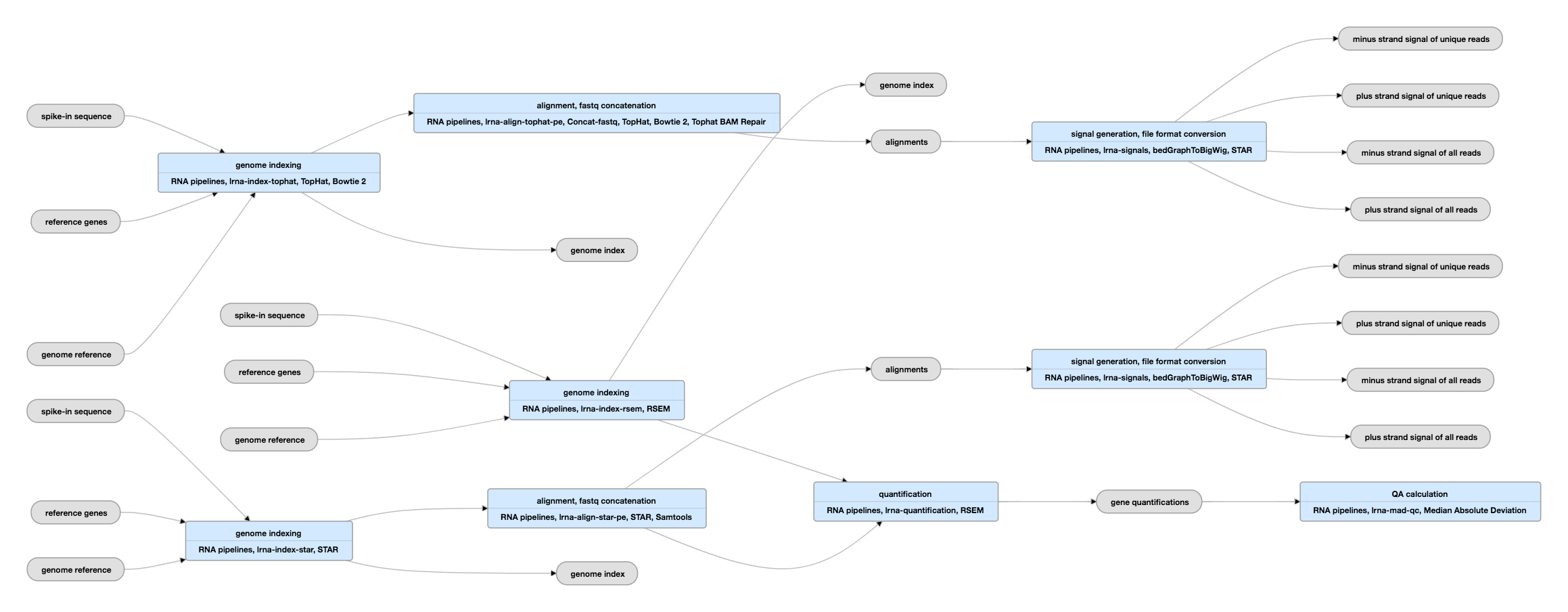
Esquema de la línea de producción para datos emparejados
Ver la instancia actual de esta línea de producción para datos emparejados
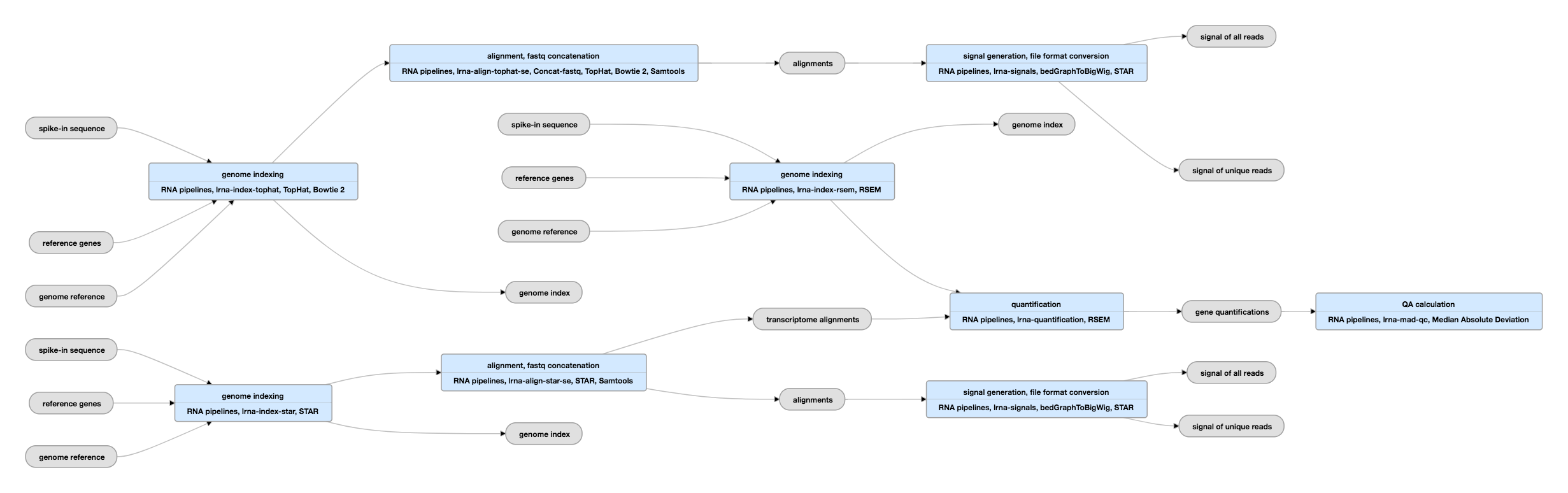
Esquema de la línea de producción para datos de un solo extremo
Ver las instancias actuales de esta línea de producción para datos de un solo extremo
Entradas:
| Formato del fichero |
Información contenida en el fichero |
Ficha descripción |
Notas |
| fastq |
lecturas |
G-zipped bulk RNA-seq reads | Las lecturas deben cumplir los criterios indicados en las Restricciones de la tubería de procesamiento uniforme. |
| tar | índice del genoma | Generado por STAR o TopHat | Por favor, vea el párrafo titulado «Con respecto a la alineación y cuantificación» debajo de la tabla «Outputs» para más información sobre los alineadores y sus índices. |
| fasta | secuencia de spike-ins | ERCC Spike-ins (External RNA Control Consortium) | Los spike-ins son efectivamente los controles para el experimento de RNA-seq. |
Salidas:
| Formato del fichero |
Información contenida en el fichero |
Descripción del fichero |
Notas |
| bam | alineaciones | Producidas por el mapeo de lecturas al genoma. | Por favor, vea el párrafo titulado «Con respecto a la alineación y la cuantificación» debajo de la tabla «Resultados» para más información sobre los alineadores y sus índices. |
| bam | alineaciones del transcriptoma | Producidas por el mapeo de lecturas al transcriptoma. | |
| bigWig | señal | Señal normalizada de RNA-seq | Para datos trenzados, las señales se generan para lecturas únicas y lecturas únicas+multimapping tanto en la cadena positiva como en la negativa. Para los datos no trenzados, se generan señales para lecturas únicas y lecturas únicas+multimapping sin tener en cuenta la identidad de la hebra. |
| tsv | cuantificaciones de genes | Incluye las cuantificaciones de picos |
Las especificaciones del formato del archivo son las siguientes:
|
| tsv | cuantificaciones de transcritos | Incluye las cuantificaciones de los spike-ins | Por favor, vea la precaución respecto a las cuantificaciones de transcritos en el párrafo de abajo titulado «Respecto a la alineación y cuantificación». |
| El pipeline también produce métricas de calidad, incluyendo la correlación de Spearman y la profundidad de lectura. | |||
Con respecto a la alineación y la cuantificación:
El mapeo de las lecturas se realiza con el programa STAR (en algunos casos, se utilizan tanto los alineadores STAR como TopHat para producir archivos bam separados) y la cuantificación de genes y transcritos se realiza con el programa RSEM. Aunque hay un acuerdo general entre los mapeos y las cuantificaciones de genes producidos por diferentes pipelines de RNA-seq, las cuantificaciones de las isoformas individuales de los transcritos, al ser mucho más complejas, pueden diferir sustancialmente dependiendo del pipeline de procesamiento empleado y son de precisión desconocida. Por lo tanto, los alineamientos y las cuantificaciones de genes pueden utilizarse con confianza, mientras que las cuantificaciones de transcritos deben usarse con cuidado.
Referencias genómicas
Ver las referencias del genoma y los tamaños de los cromosomas utilizados en este pipeline
Estos pipelines requieren tanto información de ensamblaje para la especie de interés como una referencia de genes. Cada uno de los programas principales, TopHat, STAR, y RSEM crean un índice para su uso en los pasos posteriores. Más información sobre el uso de RSEM está disponible aquí.
Controles de espiga de ARN exógeno
Los controles de espiga de ARN exógeno se añaden a las muestras para crear una línea de base estándar para la cuantificación de la expresión de ARN (PMC3166838). El consorcio ENCODE está estandarizando el uso de los controles Ambion Mix 1 disponibles en el mercado a una dilución de ~2% de las lecturas finales mapeadas. Sin embargo, hay una mezcla de datos antiguos y datos importados. Por lo tanto, para rastrear los spike-ins utilizados en una biblioteca dada, hay un conjunto de datos asociado con la biblioteca. Ese conjunto de datos contendrá el archivo de secuencia de spike-ins en formato fasta e información sobre las concentraciones. Se espera que estas secuencias de spike-ins se encuentren en el índice del genoma utilizado en el paso(s) de mapeo y en el bam generado posteriormente. Las cuantificaciones de las secuencias se pueden encontrar en los archivos de cuantificación de transcritos y genes de la RSEM.
Ver conjuntos de datos de spike-ins
Ver el certificado de análisis de los spike-ins de ERCC
Acceder al tablero de ERCC
Enlaces y publicaciones
Encontrar datos generados por este pipeline: Todos | sólo pares | sólo un extremo
Explorar publicaciones (en curso)