
- Genomi Sequencing
- Genetic Characterization
- Methods of Flu Genome Sequencing
Genome Sequencing
Influenssavirukset muuttuvat jatkuvasti, itse asiassa kaikissa influenssaviruksissa tapahtuu geneettisiä muutoksia ajan mittaan (lisätietoja on kohdassa Miten influenssavirus voi muuttua: ”Drift” ja ”Shift”). Influenssaviruksen genomi koostuu kaikista geeneistä, joista virus koostuu. CDC valvoo ympäri vuoden kiertäviä influenssaviruksia seuratakseen näiden virusten genomissa (tai genomin osissa) tapahtuvia muutoksia. Tämä työ on osa Yhdysvaltojen rutiininomaista influenssaseurantaa ja osa CDC:n roolia Maailman terveysjärjestön (WHO) influenssan viite- ja tutkimuskeskuksena (Collaborating Center for Reference and Research on Influenza). CDC:n influenssavirusten geneettisten muutosten (tunnetaan myös nimillä ”substituutiot”, ”variantit” tai ”mutaatiot”) tutkimisesta keräämillä tiedoilla on tärkeä kansanterveydellinen merkitys, sillä ne auttavat määrittämään, tehoavatko rokotteet ja viruslääkkeet nykyisin kiertäviä influenssaviruksia vastaan, sekä auttavat määrittämään, voivatko eläimissä esiintyvät influenssavirukset tarttua ihmisiin.
Geenien geenisekvensoinnilla saadaan selville nukleotidien sekvenssi, kuten aakkoset sanoissa. Nukleotidit ovat orgaanisia molekyylejä, jotka muodostavat nukleiinihappojen, kuten RNA:n tai DNA:n, rakenneyksikön rakennusosan. Kaikki influenssavirukset koostuvat yksijuosteisesta RNA:sta toisin kuin kaksijuosteinen DNA. Influenssavirusten RNA-geenit koostuvat nukleotidiketjuista, jotka on liitetty toisiinsa ja joita koodataan kirjaimilla A, C, G ja U, jotka tarkoittavat vastaavasti adeniinia, sytosiinia, guaniinia ja urasiilia. Vertaamalla yhden viruksen geenin nukleotidien koostumusta toisen viruksen geenin nukleotidien järjestykseen voidaan havaita näiden kahden viruksen välisiä vaihteluita.
Geneettiset vaihtelut ovat tärkeitä, koska ne voivat vaikuttaa influenssaviruksen pintaproteiinien rakenteeseen. Proteiinit koostuvat aminohappojen sekvensseistä.
Aminohapon korvaaminen toisella aminohapolla voi vaikuttaa viruksen ominaisuuksiin, kuten siihen, kuinka hyvin virus tarttuu ihmisten välillä ja kuinka herkkä virus on viruslääkkeille tai nykyisille rokotteille.
![]()
![]()
Genomin sekvensointi paljastaa geenin nukleotidien järjestyksen, kuten aakkoset sanoissa. Vertaamalla yhden viruksen geenin nukleotidien koostumusta toisen viruksen geenin nukleotidien järjestykseen voidaan paljastaa näiden kahden viruksen välisiä variaatioita.
Geneettiset variaatiot ovat tärkeitä, koska ne vaikuttavat influenssaviruksen pintaproteiinien rakenteeseen. Proteiinit koostuvat aminohappojen sekvensseistä.
Aminohapon korvaaminen toisella aminohapolla voi vaikuttaa viruksen ominaisuuksiin, kuten siihen, kuinka hyvin virus tarttuu ihmisten välillä ja kuinka herkkä virus on viruslääkkeille tai nykyisille rokotteille.
Influenssa A- ja B-virukset – ensisijaiset influenssavirukset, jotka tarttuvat ihmisiin – ovat RNA-viruksia, joissa on kahdeksan geenin osaa. Nämä geenit sisältävät ”ohjeet” uusien virusten tekemistä varten, ja juuri näitä ohjeita influenssavirus käyttää tartuttuaan ihmissoluun huijatakseen solua tuottamaan lisää influenssaviruksia ja siten levittämään tartuntaa.
Influenssageenit koostuvat nukleotideiksi kutsuttujen molekyylien sekvenssistä, jotka liittyvät toisiinsa ketjumaisesti. Nukleotideja merkitään kirjaimilla A, C, G ja U.
Genomin sekvensointi on prosessi, jossa määritetään nukleotidien (eli A, C, G ja U) järjestys eli sekvenssi kussakin viruksen genomissa esiintyvässä geenissä. Koko genomin sekvensoinnilla voidaan paljastaa viruksen genomin kaikkien geenien noin 13 500 kirjaimen järjestys.
Kuukausittain CDC suorittaa koko genomin sekvensoinnin noin 7 000 influenssavirukselle, jotka on saatu alkuperäisistä kliinisistä näytteistä, jotka on kerätty virologisen seurannan yhteydessä. Influenssa A- tai B-viruksen genomi sisältää kahdeksan geenisegmenttiä, jotka koodaavat (eli määrittävät viruksen 12 proteiinin rakenteen ja ominaisuudet), mukaan lukien sen kaksi ensisijaista pintaproteiinia: hemagglutiniini (HA) ja neuraminidaasi (NA). Influenssaviruksen pintaproteiinit määrittävät viruksen tärkeitä ominaisuuksia, kuten sen, miten virus reagoi tiettyihin viruslääkkeisiin, viruksen geneettisen samankaltaisuuden nykyisten influenssarokotevirusten kanssa ja zoonoottisten (eläinperäisten) influenssavirusten mahdollisuuden tartuttaa ihmisisäntiä.
Geneettinen karakterisointi
CDC ja muut kansanterveyslaboratoriot ympäri maailmaa ovat sekvensoineet influenssavirusten geenejä 1980-luvulta lähtien. CDC toimittaa geenisekvenssejä julkisiin tietokantoihin, kuten GenBankexternal icon ja Global Initiative on Sharing Avian Influenza Data (GISAID)external icon, kansanterveystutkijoiden käyttöön. Näin syntyvien geenisekvenssikirjastojen avulla CDC ja muut laboratoriot voivat verrata nykyisin kiertävien influenssavirusten geenejä vanhempien influenssavirusten ja rokotteissa käytettyjen virusten geeneihin. Tätä geenisekvenssien vertailuprosessia kutsutaan geneettiseksi karakterisoinniksi. CDC käyttää geneettistä karakterisointia seuraavista syistä:
- Määrittääkseen, kuinka läheisesti ”sukua” tai samankaltaisia influenssavirukset ovat geneettisesti keskenään
- seuratakseen, miten influenssavirukset kehittyvät
- löytääkseen geneettisiä muutoksia, jotka vaikuttavat viruksen ominaisuuksiin. Esimerkiksi sellaisten erityisten muutosten tunnistamiseksi, jotka liittyvät siihen, että influenssavirukset leviävät helpommin, aiheuttavat vakavampia tauteja tai kehittävät vastustuskykyä viruslääkkeille
- Sen arvioimiseksi, kuinka hyvin influenssarokote saattaisi suojata tiettyä influenssavirusta vastaan sen geneettisen samankaltaisuuden perusteella
- Eläinpopulaatioissa kiertävien influenssavirusten geneettisten muutosten seuraamiseksi, jotka saattaisivat antaa niille mahdollisuuden tartuttaa ihmisiä.
Influenssavirusten ryhmän väliset suhteelliset erot esitetään järjestämällä ne ”fylogeneettiseksi puuksi” kutsuttuun grafiikkaan. Influenssavirusten fylogeneettiset puut ovat kuin ihmisten sukututkimuspuut. Nämä puut osoittavat, kuinka läheistä ”sukua” yksittäiset virukset ovat toisilleen. Virukset ryhmitellään sen perusteella, ovatko niiden geenien nukleotidit identtisiä vai eivät. Influenssavirusten fylogeneettiset puut osoittavat yleensä, kuinka samankaltaisia virusten hemagglutiniini- (HA) tai neuraminidaasigeenit (NA) ovat keskenään. Jokaisella tietystä influenssaviruksesta peräisin olevalla sekvenssillä on oma haaransa puussa. Virusten välisen geneettisen eron astetta (nukleotidierojen määrää) kuvaa fylogeneettisen puun vaakasuorien viivojen (haarojen) pituus. Mitä kauempana virukset ovat toisistaan fylogeneettisen puun vaaka-akselilla, sitä enemmän virukset eroavat geneettisesti toisistaan.
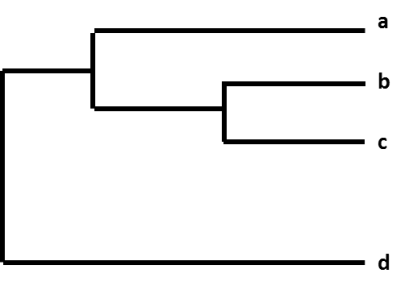
Kuva. A phylogenetic tree.
Sen jälkeen, kun esimerkiksi CDC on sekvensoinut seurannassa kerätyn influenssa A(H3N2)-viruksen, virussekvenssi luetteloidaan muiden virussekvenssien kanssa, joilla on samankaltainen HA-geeni (H3) ja samankaltainen NA-geeni (N2). Osana tätä prosessia CDC vertaa uutta virussekvenssiä muihin virussekvensseihin ja etsii niiden välisiä eroja. Tämän jälkeen CDC käyttää fylogeneettistä puuta esittämään visuaalisesti, kuinka geneettisesti A(H3N2)-virukset eroavat toisistaan.
CDC tekee influenssavirusten geneettistä karakterisointia ympäri vuoden. Näitä geneettisiä tietoja käytetään yhdessä virusten antigeenistä karakterisointia koskevien tietojen kanssa sen määrittämiseksi, mitkä rokotevirukset olisi valittava tuleviin pohjoisen tai eteläisen pallonpuoliskon influenssarokotteisiin. Helmi- ja syyskuussa pidettäviä WHO:n rokotekonsultointikokouksia edeltävinä kuukausina CDC kerää influenssaviruksia seurannan avulla ja vertaa nykyisten rokotevirusten HA- ja NA-geenisekvenssejä kiertävien influenssavirusten vastaaviin. Tämä on yksi tapa arvioida, kuinka läheistä sukua kiertävät influenssavirukset ovat viruksille, joita vastaan kausi-influenssarokote on suunniteltu suojaamaan. Kun viruksia kerätään ja karakterisoidaan geneettisesti, eroja voi paljastua.
Joskus esimerkiksi kauden aikana kiertävät virukset muuttuvat geneettisesti, jolloin ne eroavat vastaavasta rokoteviruksesta. Tämä on yksi osoitus siitä, että seuraavan influenssakauden rokotetta varten on ehkä valittava erilainen rokotevirus, vaikka muut tekijät, mukaan lukien antigeenisen karakterisoinnin löydökset, vaikuttavat voimakkaasti rokotepäätöksiin. Influenssavirusten HA- ja NA-pintaproteiinit ovat antigeenejä, mikä tarkoittaa, että immuunijärjestelmä tunnistaa ne ja ne pystyvät laukaisemaan immuunivasteen, mukaan luettuna vasta-aineiden tuotanto, joka voi estää infektion. Antigeeninen karakterisointi tarkoittaa viruksen vasta-aineiden kanssa tapahtuvan reaktion analysointia, jonka avulla voidaan arvioida sen suhdetta toiseen virukseen.
Flunssan genomin sekvensointimenetelmät
Yksi influenssanäyte sisältää monia koeputkessa kasvatettuja influenssaviruksen hiukkasia, joiden geneettiset eroavaisuudet ovat usein vähäisiä verrattuna toisiinsa koko sisarviruspopulaatiosta.
Traditionaalisesti tutkijat ovat käyttäneet ”Sangerin reaktioksi” kutsuttua sekvensointitekniikkaa influenssan evoluution seuraamiseen osana virologista seurantaa. Sangerin sekvensoinnilla identifioidaan vallitseva geneettinen sekvenssi isolaatissa esiintyvien monien influenssavirusten joukosta. Tämä tarkoittaa, että näytteessä olevien virusten populaatiossa esiintyvät pienet vaihtelut eivät näy lopullisessa tuloksessa. Tutkijat käyttävät Sanger-menetelmää usein influenssavirusten osittaiseen genomin sekvensointiin, kun taas uudemmat tekniikat (ks. seuraava kohta) soveltuvat paremmin koko genomin sekvensointiin.
Viiden viime vuoden aikana CDC on käyttänyt ”seuraavan sukupolven sekvensointimenetelmiä” (Next Generation Sequencing (NGS)), jotka ovat lisänneet huomattavasti sekvensointianalyysin avulla saatavan tiedon määrää ja yksityiskohtaisuutta. NGS käyttää kehittynyttä molekyylitunnistusta (Advanced Molecular Detection, AMD) kunkin näytteessä olevan viruksen geenisekvenssien tunnistamiseen. Näin ollen NGS paljastaa geneettiset vaihtelut monien eri influenssavirushiukkasten välillä yhdessä näytteessä, ja nämä menetelmät paljastavat myös genomien koko koodaavan alueen. Tällainen yksityiskohtaisuus voi suoraan hyödyttää kansanterveydellistä päätöksentekoa tärkeillä tavoilla, mutta korkeasti koulutettujen asiantuntijoiden on tulkittava tietoja huolellisesti muiden saatavilla olevien tietojen yhteydessä. Katso AMD-hankkeet: Lisätietoa siitä, miten NGS ja AMD mullistavat influenssagenomikartoituksen CDC:ssä.