
Survol du pipeline
Le pipeline RNA-seq en vrac a été développé dans le cadre de la série de pipelines de traitement uniformes ENCODE. Le code complet du pipeline est librement disponible sur Github et peut être exécuté sur DNAnexus (le lien nécessite la création d’un compte) à leur tarif actuel.
Le pipeline ENCODE Bulk RNA-seq peut être utilisé pour les bibliothèques d’ARN-seq répliquées et non répliquées, appariées ou à extrémité unique, et spécifiques ou non à un brin. Les bibliothèques doivent être générées à partir d’ARNm (poly(A)+, ARN total appauvri en ARNr, ou populations poly(A)- qui sont sélectionnées par taille pour être plus longues qu’environ 200 pb. À l’avenir, ce pipeline pourra également être utilisé pour traiter les données PAS-seq et Bru-seq.
Schéma de pipeline pour les données appariées
Voir l’instance actuelle de ce pipeline pour les données appariées
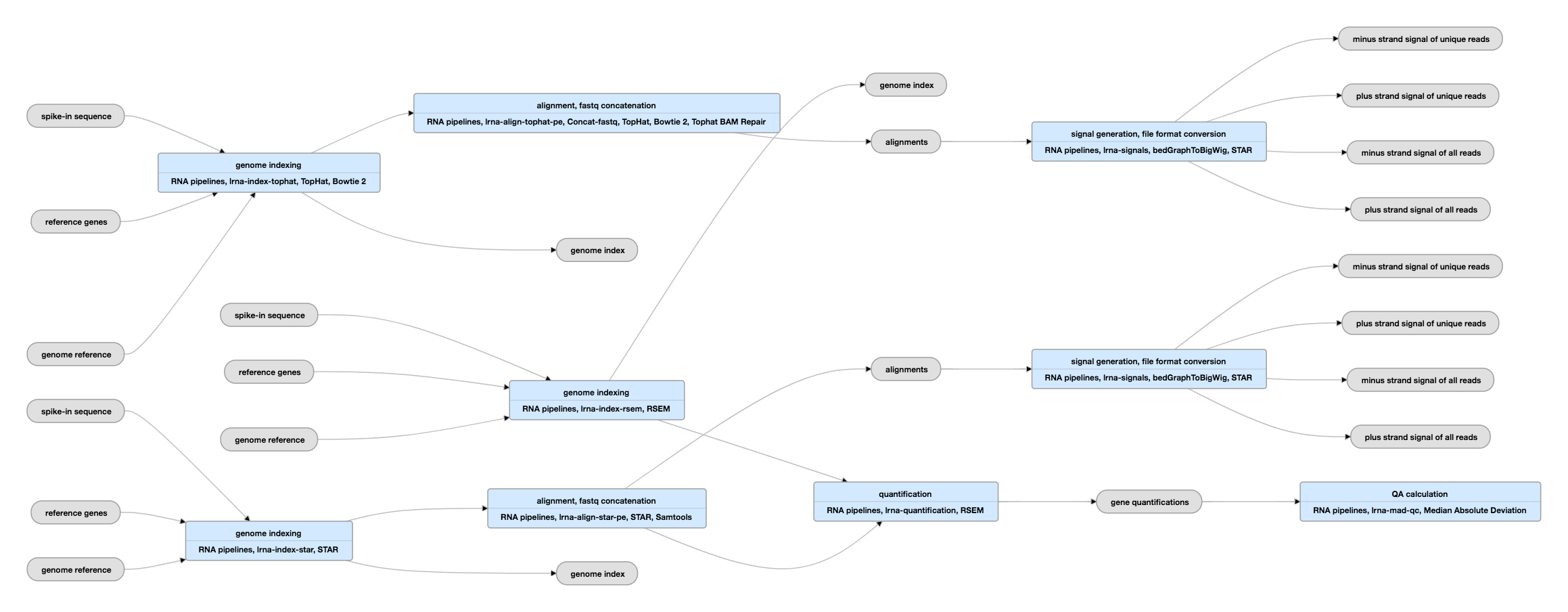
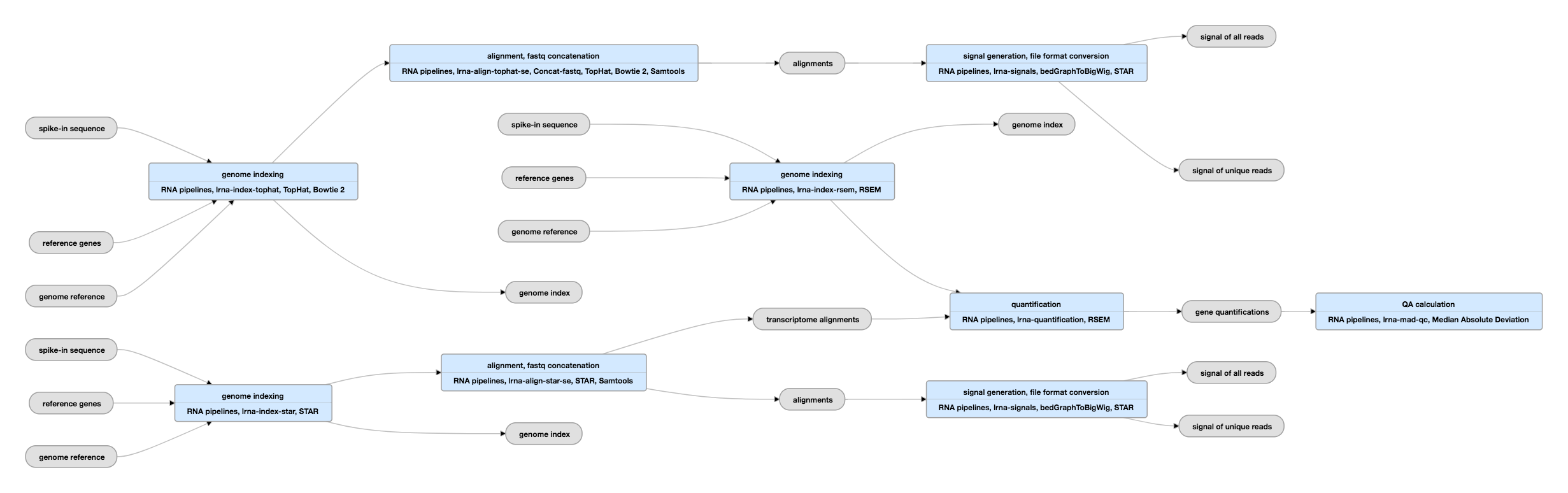
Schéma de pipeline pour les données à extrémité unique
Voir les instances actuelles de ce pipeline pour les données à extrémité unique
Entrées :
| Format du fichier |
Information contenue dans le fichier |
Fichier. description |
Notes |
| fastq |
Lectures |
G-lectures RNA-seq en vrac zippées | Les lectures doivent répondre aux critères décrits dans les restrictions du pipeline de traitement uniforme. |
| tar | Indice génomique | Généré par STAR ou TopHat | Veuillez consulter le paragraphe intitulé « Concernant l’alignement et la quantification » sous le tableau « Sorties » pour en savoir plus sur les aligneurs et leurs indices. |
| fasta | séquence spike-in | Spike-ins ERCC (External RNA Control Consortium) | Les spike-ins sont effectivement les contrôles de l’expérience RNA-seq. |
Sorties :
| Format du fichier |
Information contenue dans le fichier |
Description du fichier |
Notes |
| bam | alignements | Produit par la mise en correspondance des lectures avec le génome. | Veuillez consulter le paragraphe intitulé « Concernant l’alignement et la quantification » sous le tableau « Sorties » pour en savoir plus sur les aligneurs et leurs indices. |
| bam | alignements transcriptomiques | Produits par la mise en correspondance des lectures avec le transcriptome. | |
| bigWig | signal | Signal RNA-seq normalisé | Pour les données brinées, les signaux sont générés pour les lectures uniques et les lectures uniques+multimapping dans les deux brins plus et moins. Pour les données non brins, les signaux sont générés pour les lectures uniques et les lectures uniques+multimapping sans tenir compte de l’identité du brin. |
| tsv | quantifications de gènes | Inclut les quantifications de spike-ins |
Les spécifications du format de fichier sont les suivantes:
|
| tsv | quantifications de transcriptions | Inclut les quantifications de spike-ins | Veuillez voir la mise en garde concernant les quantifications de transcriptions dans le paragraphe ci-dessous intitulé « Concernant l’alignement et la quantification ». |
| Le pipeline produit également des métriques de qualité, notamment la corrélation de Spearman et la profondeur de lecture. | |||
En ce qui concerne l’alignement et la quantification:
La cartographie des lectures est effectuée à l’aide du programme STAR (dans certains cas, les aligneurs STAR et TopHat sont utilisés pour produire des fichiers bam distincts) et la quantification des gènes et des transcrits est effectuée à l’aide du programme RSEM. Bien qu’il y ait une concordance générale entre les alignements et les quantifications des gènes produits par différents pipelines d’ARN séquentiel, les quantifications des isoformes de transcription individuelles, beaucoup plus complexes, peuvent différer considérablement selon le pipeline de traitement utilisé et leur précision est inconnue. Par conséquent, les alignements et les quantifications de gènes peuvent être utilisés avec confiance, tandis que les quantifications de transcriptions doivent être utilisées avec précaution.
Références génomiques
Voir les références génomiques et les tailles de chromosomes utilisées dans ce pipeline
Ces pipelines nécessitent à la fois des informations d’assemblage pour l’espèce d’intérêt et une référence génique. Chacun des principaux programmes, TopHat, STAR et RSEM, crée un index à utiliser dans les étapes suivantes. Plus d’informations sur l’utilisation de RSEM sont disponibles ici.
Contrôles de spike-in d’ARN exogène
Des contrôles de spike-in d’ARN exogène sont ajoutés aux échantillons pour créer une ligne de base standard pour la quantification de l’expression de l’ARN (PMC3166838). Le consortium ENCODE standardise l’utilisation des spike-ins Ambion Mix 1 disponibles dans le commerce à une dilution de ~2% des lectures finales cartographiées. Cependant, il existe un mélange de données anciennes et de données importées. Par conséquent, pour suivre les spike-ins utilisés dans une bibliothèque donnée, il existe un jeu de données associé à la bibliothèque. Ce jeu de données contient le fichier de séquence des spike-ins au format fasta et des informations sur les concentrations. Ces séquences de spike-ins sont censées se trouver dans l’index du génome utilisé dans la ou les étapes de cartographie et dans le bam généré par la suite. Les quantifications des séquences se trouvent dans les fichiers de quantification des transcriptions et des gènes RSEM.
Voir les ensembles de données spike-ins
Voir le certificat d’analyse pour les spike-ins ERCC
Accéder au tableau de bord ERCC
Liens et publications
Trouver les données générées par ce pipeline : Toutes | paired-end seulement | single-end seulement
Explorer les publications (en cours)
.