
A csővezeték áttekintése
A Bulk RNA-seq csővezetéket az ENCODE Uniform Processing Pipelines sorozat részeként fejlesztették ki. A teljes csővezeték kódja szabadon elérhető a Githubon, és a DNAnexuson futtatható (a linkhez fiók létrehozása szükséges) a jelenlegi árakon.
Az ENCODE Bulk RNA-seq pipeline replikált és nem replikált, párosított vagy egyvégű, valamint szálspecifikus vagy nem szálspecifikus RNA-seq könyvtárakhoz egyaránt használható. A könyvtárakat mRNS-ből (poli(A)+, rRNS-mentesített teljes RNS-ből vagy poli(A)- populációkból kell létrehozni, amelyek mérete úgy van szelektálva, hogy körülbelül 200 bp-nél hosszabbak legyenek. A jövőben ez a csővezeték a PAS-seq és Bru-seq adatok feldolgozására is használható lesz.
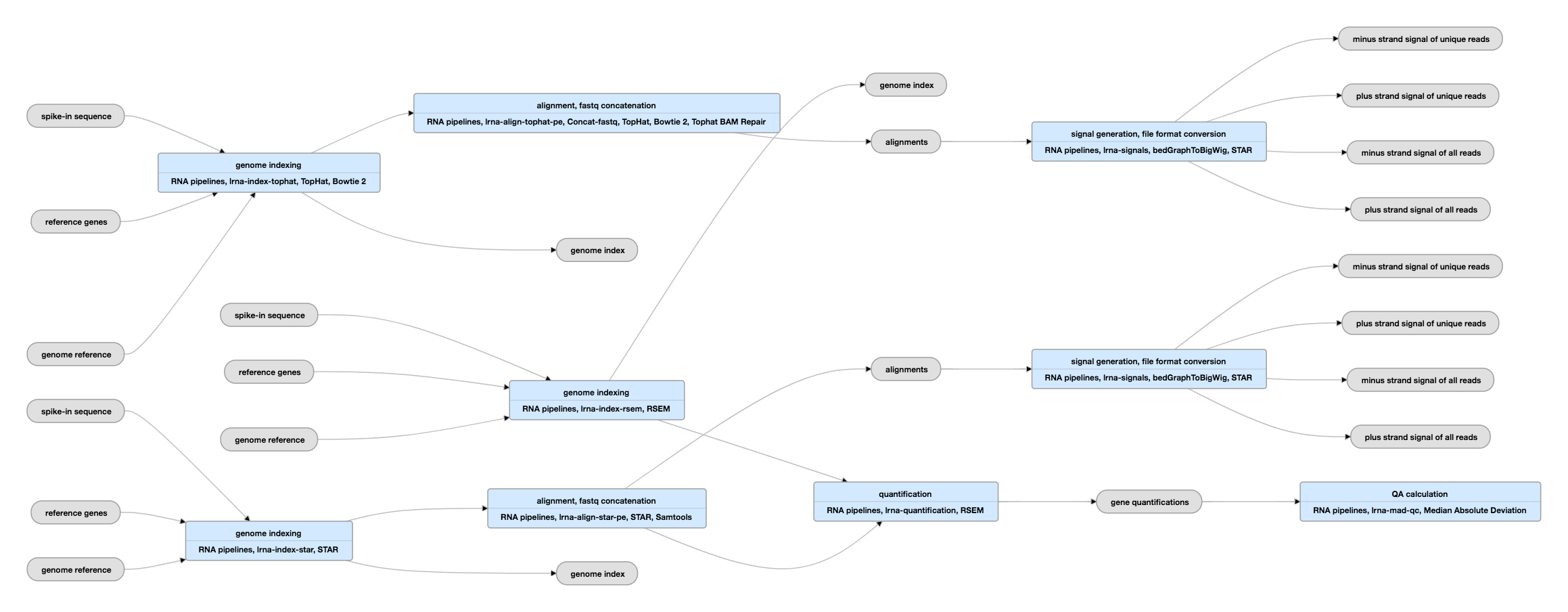
Pipeline sémája párosított végű adatokhoz
Nézze meg a pipeline aktuális példányát párosított végű adatokhoz
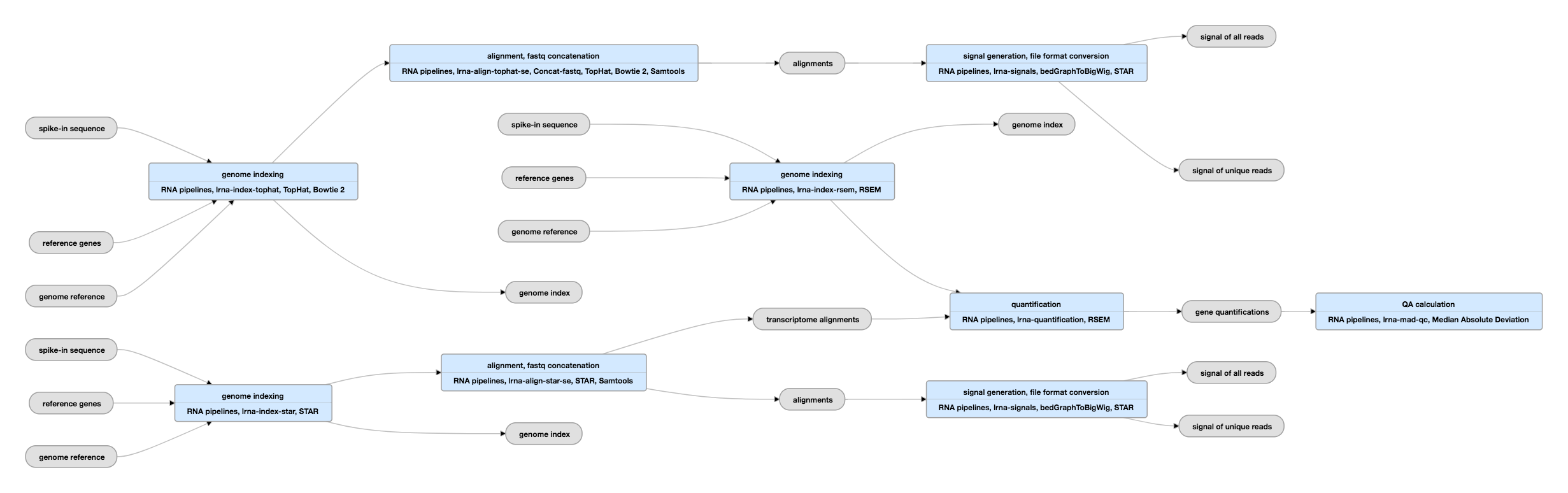
Pipeline sémája egyvégű adatokhoz
Nézze meg a pipeline aktuális példányait egyvégű adatokhoz
Bemenetek:
| Fájlformátum |
A fájlban található információ |
Fájl. description |
Notes |
| fastq |
reads |
G-zippelt tömeges RNS-seq leolvasások | A leolvasásoknak meg kell felelniük az Uniform Processing Pipeline Restrictions (Egységes feldolgozási pipeline korlátozások) című dokumentumban meghatározott kritériumoknak. |
| tar | genom index | Generated by STAR or TopHat | Az “Outputs” táblázat alatti “Regarding alignment and quantification” című bekezdésben olvasható bővebben az alignerekről és indexeikről. |
| fasta | spike-in szekvencia | ERCC Spike-in (External RNA Control Consortium) | A spike-inek gyakorlatilag az RNS-seq kísérlet kontrolljai. |
Kimenetek:
| Fájlformátum |
A fájlban szereplő információk |
Fájlleírás |
Jegyzetek |
| bam | alignments | A leolvasások genomra való leképezésével keletkezik. | Az “Outputs” táblázat alatti “Regarding alignment and quantification” című bekezdésben olvasható az alignerekről és indexeikről. |
| bam | transcriptome alignments | Produced by mapping reads to the transcriptome. | |
| bigWig | signal | Normalized RNA-seq signal | Stranded adatok esetén a jeleket az egyedi olvasatok és az egyedi+multimapping olvasatok számára generáljuk mind a plusz, mind a mínusz szálon. Szál nélküli adatok esetén a jeleket az egyedi leolvasásokra és az egyedi+multimapping leolvasásokra generáljuk, a szálazonosságra való tekintet nélkül. |
| tsv | gének mennyiségi meghatározása | Tartalmazza a spike-ins mennyiségi meghatározásokat |
A fájlformátum specifikációja a következő:
|
| tsv | transcript quantifications | Includes the spike-ins quantifications | Kérem, olvassa el a “Regarding alignment and quantification” című bekezdésben a transzkriptek mennyiségi meghatározásával kapcsolatos figyelmeztetést. |
| A csővezeték minőségi metrikákat is készít, beleértve a Spearman-korrelációt és az olvasási mélységet. | |||
Az igazítással és a mennyiségi meghatározással kapcsolatban:
A leolvasások leképezése a STAR programmal történik (egyes esetekben mind a STAR, mind a TopHat igazítót használják, hogy külön bam fájlokat készítsenek), a gének és transzkriptek mennyiségi meghatározása pedig az RSEM programmal történik. Bár a különböző RNS-seq-pipeline-ok által előállított leképezések és génkvantitások között általános egyetértés van, az egyes transzkript izoformák kvantitatív meghatározása, mivel sokkal összetettebb, jelentősen eltérhet az alkalmazott feldolgozási pipeline-tól függően, és pontossága nem ismert. Ezért az illesztések és a génkvantitások magabiztosan használhatók, míg a transzkriptek kvantitásait óvatosan kell használni.
Genomikus referenciák
Nézze meg az ebben a pipeline-ban használt genom-referenciákat és kromoszómaméreteket
Ezeknek a pipelineknek mind az adott fajra vonatkozó összeállítási információkra, mind pedig egy génreferenciára van szükségük. A fő programok, a TopHat, a STAR és az RSEM mindegyike létrehoz egy indexet a későbbi lépésekben való felhasználáshoz. Az RSEM használatáról további információ itt található.
Exogén RNS spike-in kontrollok
A mintákhoz exogén RNS spike-in kontrollokat adunk, hogy standard alapvonalat hozzunk létre az RNS-expresszió mennyiségi meghatározásához (PMC3166838). Az ENCODE konzorcium szabványosítja a kereskedelmi forgalomban kapható Ambion Mix 1 spike-inek használatát a végső leképezett olvasatok ~2%-ának megfelelő hígításban. A régebbi adatok és az importált adatok azonban keverednek. Ezért egy adott könyvtárban használt spike-inek nyomon követésére van egy, a könyvtárhoz kapcsolódó adatkészlet. Ez az adatkészlet tartalmazza a spike-ins szekvenciafájlt fasta formátumban és a koncentrációkra vonatkozó információkat. Ezek a spike-in szekvenciák várhatóan megtalálhatók a térképezési lépés(ek)ben használt genomindexben és a később generált bam-ben. A szekvenciák mennyiségi meghatározása megtalálható az RSEM transzkript és gén mennyiségi meghatározási fájlokban.
Nézze meg a spike-ins adatkészleteket
Nézze meg az ERCC spike-ins elemzési tanúsítványát
Lépjen be az ERCC dash boardra
Linkek és publikációk
Keresje meg az e csővezeték által generált adatokat:
Közlemények keresése (folyamatban lévő)