
- Genom Szekvenálás
- Genetikai jellemzés
- Az influenza genom szekvenálás módszerei
Genom szekvenálás
Az influenzavírusok folyamatosan változnak, valójában minden influenzavírus idővel genetikai változásokon megy keresztül (további információért lásd: Hogyan változhat az influenzavírus: “Drift” és “Shift”). Az influenzavírus genomja a vírust alkotó összes génből áll. A CDC egész évben felügyeli a forgalomban lévő influenzavírusokat, hogy nyomon kövesse e vírusok genomjának (vagy genomrészleteinek) változásait. Ezt a munkát a rutinszerű amerikai influenza-felügyelet részeként és a CDC-nek az Egészségügyi Világszervezet (WHO) influenzaügyi referencia- és kutatási együttműködő központjaként betöltött szerepének részeként végzik. A CDC által az influenzavírusok genetikai változásainak (más néven “helyettesítések”, “variánsok” vagy “mutációk”) tanulmányozásából gyűjtött információk fontos közegészségügyi szerepet játszanak, mivel segítenek meghatározni, hogy a vakcinák és a vírusellenes gyógyszerek működnek-e a jelenleg keringő influenzavírusok ellen, valamint segítenek meghatározni, hogy az állatokban lévő influenzavírusok megfertőzhetik-e az embert.
A génszekvenálás feltárja a nukleotidok sorrendjét egy génben, mint az ábécé betűi a szavakban. A nukleotidok olyan szerves molekulák, amelyek a nukleinsavak, például az RNS vagy a DNS szerkezeti egységét alkotják. Minden influenzavírus egyszálú RNS-ből áll, szemben a kétszálú DNS-sel. Az influenzavírusok RNS-génjei nukleotidok láncaiból állnak, amelyek egymáshoz kapcsolódnak, és amelyeket az A, C, G és U betűk kódolnak, amelyek az adenint, citozint, guanint és uracilt jelentik. Ha összehasonlítjuk a nukleotidok összetételét egy vírus génjében és a nukleotidok sorrendjét egy másik vírus génjében, akkor a két vírus között eltéréseket fedezhetünk fel.
A genetikai eltérések azért fontosak, mert befolyásolhatják az influenzavírus felszíni fehérjéinek szerkezetét. A fehérjék aminosavak szekvenciáiból állnak.
Az egyik aminosav cseréje egy másikra befolyásolhatja a vírus tulajdonságait, például azt, hogy a vírus mennyire jól terjed az emberek között, és mennyire fogékony a vírus a vírusellenes gyógyszerekre vagy a jelenlegi vakcinákra.
![]()
![]()
A génszekvenálás feltárja a nukleotidok sorrendjét egy génben, mint az ábécé betűi a szavakban. Ha összehasonlítjuk a nukleotidok összetételét egy vírus génjében és a nukleotidok sorrendjét egy másik vírus génjében, felfedezhetjük a két vírus közötti eltéréseket.
A genetikai eltérések azért fontosak, mert befolyásolják az influenzavírus felszíni fehérjéinek szerkezetét. A fehérjék aminosavak szekvenciáiból állnak.
Az egyik aminosav cseréje egy másikra befolyásolhatja a vírus tulajdonságait, például azt, hogy a vírus mennyire jól terjed az emberek között, és mennyire fogékony a vírus a vírusellenes gyógyszerekre vagy a jelenlegi vakcinákra.
Az influenza A és B vírusok – az embereket megfertőző elsődleges influenzavírusok – RNS-vírusok, amelyek nyolc génszakaszból állnak. Ezek a gének tartalmazzák az új vírusok előállítására vonatkozó “utasításokat”, és ezeket az utasításokat használja az influenzavírus, amint megfertőz egy emberi sejtet, hogy rávegye a sejtet további influenzavírusok előállítására, ezáltal terjesztve a fertőzést.
Az influenzavírus génjei nukleotidoknak nevezett molekulák sorozatából állnak, amelyek láncszerű formában kapcsolódnak egymáshoz. A nukleotidokat A, C, G és U betűkkel jelölik.
A génszekvenálás olyan folyamat, amely meghatározza a vírus genomjában található egyes génekben lévő nukleotidok (azaz A, C, G és U) sorrendjét, vagyis szekvenciáját. A teljes genomszekvenálással a vírus genomjának valamennyi génjének körülbelül 13 500 betűből álló szekvenciája feltárható.
A CDC minden évben körülbelül 7000 influenzavírus teljes genomszekvenálását végzi el a virológiai felügyelet keretében gyűjtött eredeti klinikai mintákból. Az influenza A vagy B vírus genomja nyolc génszakaszt tartalmaz, amelyek kódolják (azaz meghatározzák a vírus 12 fehérjéjének szerkezetét és jellemzőit), beleértve a két elsődleges felszíni fehérjét: a hemagglutinint (HA) és a neuraminidázt (NA). Az influenzavírus felszíni fehérjéi határozzák meg a vírus fontos tulajdonságait, beleértve azt, hogy a vírus hogyan reagál bizonyos vírusellenes gyógyszerekre, a vírus genetikai hasonlóságát a jelenlegi influenza vakcinavírusokkal, valamint a zoonózisos (állati eredetű) influenzavírusok emberi gazdaszervezetek megfertőzésének lehetőségét.
Genetikai jellemzés
A CDC és más közegészségügyi laboratóriumok világszerte az 1980-as évek óta szekvenálják az influenzavírusok génjeit. A CDC hozzájárul a génszekvenciákhoz nyilvános adatbázisokhoz, mint például a GenBankexkülső ikon és a Global Initiative on Sharing Avian Influenza Data (GISAID)külső ikon, a közegészségügyi kutatók általi felhasználás céljából. Az így létrejövő génszekvencia-könyvtárak lehetővé teszik a CDC és más laboratóriumok számára, hogy összehasonlítsák a jelenleg forgalomban lévő influenzavírusok génjeit a régebbi influenzavírusok és a vakcinákban használt vírusok génjeivel. A genetikai szekvenciák összehasonlításának ezt a folyamatát genetikai jellemzésnek nevezik. A CDC a genetikai jellemzést a következő okokból használja:
- Meghatározni, hogy az influenzavírusok genetikailag mennyire “rokonok” vagy hasonlóak egymáshoz
- megfigyelni, hogyan fejlődnek az influenzavírusok
- azonosítani a vírus tulajdonságait befolyásoló genetikai változásokat. Például azon konkrét változások azonosítása, amelyek az influenzavírusok könnyebb terjedésével, súlyosabb betegséget okozó vagy a vírusellenes gyógyszerekkel szembeni rezisztencia kialakulásával kapcsolatosak
- Az influenzavírus és a vírus genetikai hasonlósága alapján annak felmérése, hogy egy influenza elleni védőoltás mennyire véd egy adott influenzavírus ellen
- Az állatpopulációkban keringő influenzavírusok olyan genetikai változásainak megfigyelése, amelyek lehetővé tehetik számukra az emberek megfertőzését.
Az influenzavírusok egy csoportja közötti relatív különbségeket egy “filogenetikai fának” nevezett grafikonba rendezve mutatják be. Az influenzavírusok filogenetikai fái olyanok, mint az emberek családfái (genealógia). Ezek a fák megmutatják, hogy az egyes vírusok milyen szoros “rokonságban” állnak egymással. A vírusokat aszerint csoportosítják, hogy génjeik nukleotidjai azonosak-e vagy sem. Az influenzavírusok filogenetikai fái általában azt mutatják, hogy a vírusok hemagglutinin (HA) vagy neuraminidáz (NA) génjei mennyire hasonlítanak egymáshoz. Egy adott influenzavírus minden egyes szekvenciája saját ággal rendelkezik a fán. A vírusok közötti genetikai különbség mértékét (a nukleotidkülönbségek számát) a filogenetikai fán a vízszintes vonalak (ágak) hossza mutatja. Minél távolabb vannak egymástól a vírusok a filogenetikai fa vízszintes tengelyén, annál jobban különböznek egymástól genetikailag a vírusok.
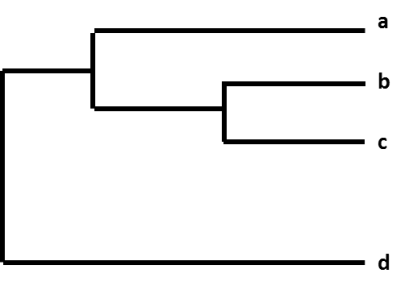
Ábra. Egy filogenetikai fa.
A CDC például egy megfigyelés során begyűjtött influenza A(H3N2) vírus szekvenálása után a vírusszekvenciát más, hasonló HA génnel (H3) és hasonló NA génnel (N2) rendelkező vírusszekvenciákkal együtt katalogizálja. Ennek a folyamatnak a részeként a CDC összehasonlítja az új vírusszekvenciát a többi vírusszekvenciával, és különbségeket keres közöttük. A CDC ezután egy filogenetikai fa segítségével vizuálisan ábrázolja, hogy az A(H3N2) vírusok genetikailag mennyire különböznek egymástól.
A CDC egész évben végzi az influenzavírusok genetikai jellemzését. Ezeket a genetikai adatokat a vírusok antigénjellemzési adataival együtt használják fel annak meghatározásához, hogy mely vakcinavírusokat kell kiválasztani a közelgő északi vagy déli féltekei influenza elleni vakcinákhoz. A WHO februári és szeptemberi vakcinával kapcsolatos konzultációs üléseit megelőző hónapokban a CDC megfigyelés útján összegyűjti az influenzavírusokat, és összehasonlítja a jelenlegi vakcinavírusok HA- és NA-génszekvenciáit a forgalomban lévő influenzavírusokéval. Ez az egyik módja annak felmérésére, hogy a keringő influenzavírusok mennyire állnak közeli rokonságban azokkal a vírusokkal, amelyek ellen a szezonális influenza elleni vakcina védelmet nyújt. Ahogy a vírusokat összegyűjtik és genetikailag jellemzik, feltárhatók a különbségek.
Egy szezon során például néha a keringő vírusok genetikailag megváltoznak, ami miatt eltérnek a megfelelő vakcinavírustól. Ez az egyik jele annak, hogy a következő influenzaszezon vakcinájához esetleg más vakcinavírust kell választani, bár más tényezők, többek között az antigénjellemzési eredmények is nagyban befolyásolják a vakcinával kapcsolatos döntéseket. Az influenzavírusok HA és NA felszíni fehérjéi antigének, ami azt jelenti, hogy az immunrendszer felismeri őket, és képesek immunválaszt kiváltani, beleértve a fertőzést blokkolni képes antitestek termelődését. Az antigénjellemzés egy vírus antitestekkel való reakciójának elemzésére utal, hogy segítsen felmérni, hogyan viszonyul egy másik vírushoz.
Az influenza genom szekvenálásának módszerei
Egy influenzaminta sok, kémcsőben tenyésztett influenzavírusrészecskét tartalmaz, amelyek gyakran kis genetikai különbségekkel rendelkeznek egymáshoz képest a testvérvírusok teljes populációjában.
A tudósok hagyományosan a “Sanger-reakciónak” nevezett szekvenálási technikát használják az influenza evolúciójának nyomon követésére a virológiai felügyelet részeként. A Sanger-szekvenálás azonosítja a domináns genetikai szekvenciát az izolátumban található számos influenzavírus között. Ez azt jelenti, hogy a mintában jelen lévő víruspopuláció apró eltérései nem tükröződnek a végeredményben. A tudósok gyakran használják a Sanger-módszert az influenzavírusok részleges genomszekvenálására, míg az újabb technológiák (lásd a következő bekezdést) jobban alkalmasak a teljes genom szekvenálására.
A CDC az elmúlt öt évben az “újgenerációs szekvenálás (NGS)” módszereit alkalmazta, amelyek jelentősen megnövelték a szekvenáló elemzés által szolgáltatható információk mennyiségét és részletességét. Az NGS fejlett molekuláris detektálást (AMD) használ a mintában lévő egyes vírusok génszekvenciáinak azonosítására. Ezért az NGS feltárja az egyetlen mintában lévő sok különböző influenzavírusrészecske közötti genetikai eltéréseket, és ezek a módszerek a genomok teljes kódoló régióját is feltárják. Ez a részletesség közvetlenül és fontos módon segítheti a közegészségügyi döntéshozatalt, de az adatokat magasan képzett szakértőknek kell gondosan értelmezniük a rendelkezésre álló egyéb információk összefüggésében. Lásd: AMD projektek: Az NGS és az AMD forradalmasítja az influenza genom feltérképezését a CDC-nél.