
Panoramica della pipeline
La Bulk RNA-seq pipeline è stata sviluppata come parte della serie ENCODE Uniform Processing Pipelines. Il codice completo della pipeline è liberamente disponibile su Github e può essere eseguito su DNAnexus (il link richiede la creazione di un account) al loro prezzo attuale.
La pipeline ENCODE Bulk RNA-seq può essere utilizzata sia per le librerie RNA-seq replicate e non replicate, paired-ended o single-ended, e strand-specific o non strand-specific. Le librerie devono essere generati da mRNA (poly (A) +, rRNA-depleted RNA totale, o poly (A) – popolazioni che sono dimensioni-selezionati per essere più lungo di circa 200 bp. In futuro, questa pipeline può anche essere utilizzata per elaborare i dati PAS-seq e Bru-seq.
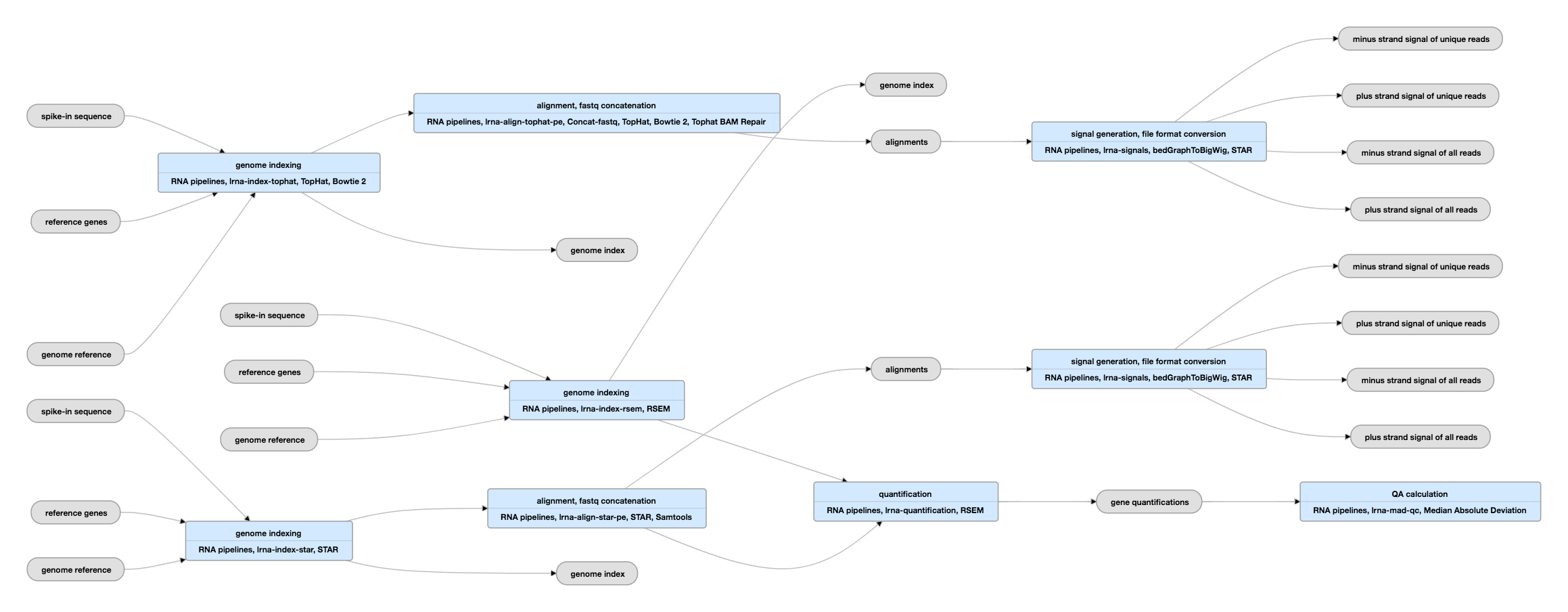
Schema della pipeline per dati appaiati
Vedi l’istanza corrente di questa pipeline per dati appaiati
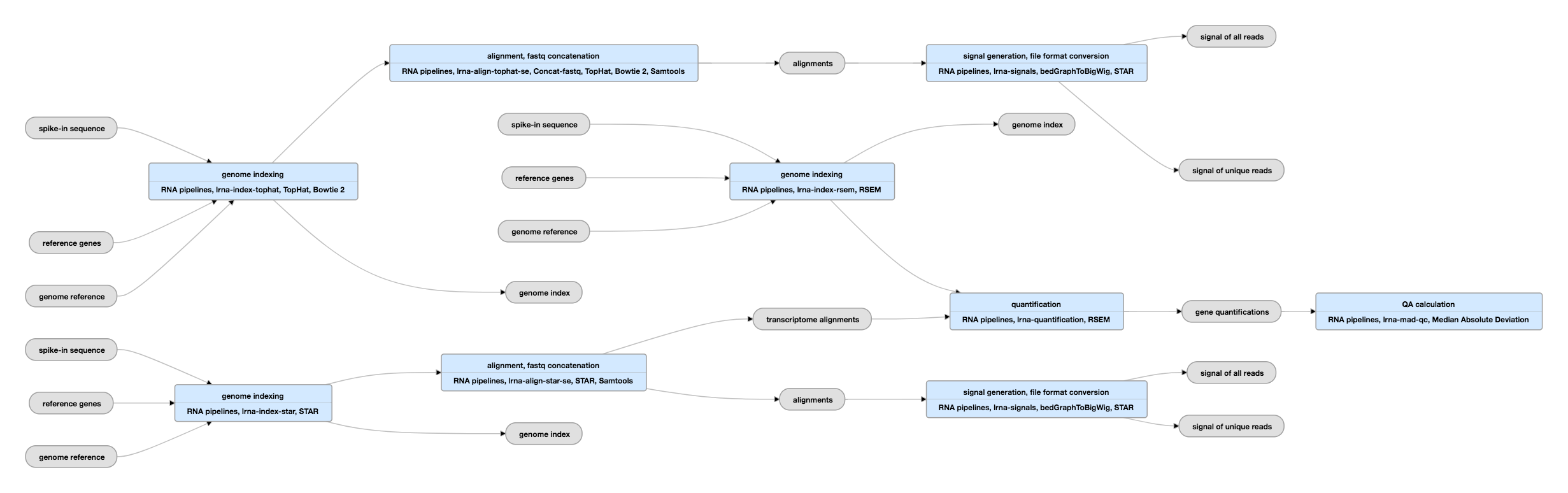
Schema della pipeline per dati single-ended
Vedi le istanze correnti di questa pipeline per dati single-ended
Input:
| Formato del file |
Informazioni contenute nel file |
File descrizione |
Note |
| fastq |
leggi |
G-Letture di RNA-seq in blocco zippate | Le letture devono soddisfare i criteri delineati nelle Restrizioni della pipeline di elaborazione uniforme. |
| tar | indice del genoma | Generato da STAR o TopHat | Per favore vedi il paragrafo intitolato “Riguardo l’allineamento e la quantificazione” sotto la tabella “Outputs” per maggiori informazioni sugli allineatori e i loro indici. |
| fasta | sequenza spike-in | ERCC Spike-ins (External RNA Control Consortium) | Gli spike-ins sono effettivamente i controlli dell’esperimento RNA-seq. |
Output:
| Formato del file |
Informazioni contenute nel file |
Descrizione del file |
Note |
| bam | allineamenti | Prodotti dalla mappatura delle letture al genoma. | Si prega di vedere il paragrafo intitolato “Per quanto riguarda l’allineamento e la quantificazione” sotto la tabella “Outputs” per ulteriori informazioni sugli allineatori e i loro indici. |
| bam | allineamenti del trascrittoma | Prodotti dalla mappatura delle letture al trascrittoma. | |
| bigWig | segnale | Segnale normalizzato RNA-seq | Per i dati a filamento, i segnali sono generati per le letture uniche e le letture uniche+multimappatura in entrambi i filamenti positivo e negativo. Per i dati non filiformi, i segnali sono generati per le letture uniche e le letture uniche+multimappatura senza considerare l’identità del filamento. |
| tsv | quantificazioni geniche | Include le quantificazioni spike-ins |
Le specifiche del formato del file sono le seguenti:
|
| tsv | quantificazioni di trascrizione | Include le quantificazioni degli spike-in | Si prega di vedere l’avvertimento riguardante le quantificazioni di trascrizione nel paragrafo sottostante intitolato “Regarding alignment and quantification”. |
| La pipeline produce anche metriche di qualità, tra cui la correlazione Spearman e la profondità di lettura. | |||
Per quanto riguarda l’allineamento e la quantificazione:
La mappatura delle letture è fatta usando il programma STAR (in alcuni casi, entrambi gli allineatori STAR e TopHat sono usati per produrre file bam separati) e la quantificazione di geni e trascrizioni è fatta con il programma RSEM. Sebbene ci sia un accordo generale tra le mappature e le quantificazioni dei geni prodotte da diverse pipeline di RNA-seq, le quantificazioni delle singole isoforme di trascrizione, essendo molto più complesse, possono differire sostanzialmente a seconda della pipeline di elaborazione impiegata e sono di precisione sconosciuta. Pertanto, gli allineamenti e le quantificazioni dei geni possono essere usati con fiducia, mentre le quantificazioni dei trascritti dovrebbero essere usate con cautela.
Riferimenti genomici
Vedi i riferimenti del genoma e le dimensioni dei cromosomi usati in questa pipeline
Queste pipeline richiedono sia informazioni di assemblaggio per le specie di interesse che un riferimento genico. Ognuno dei programmi principali, TopHat, STAR e RSEM crea un indice da usare nei passi successivi. Maggiori informazioni sull’uso di RSEM sono disponibili qui.
Controlli spike-in di RNA esogeno
I controlli spike-in di RNA esogeno vengono aggiunti ai campioni per creare una linea di base standard per la quantificazione dell’espressione di RNA (PMC3166838). Il consorzio ENCODE si sta standardizzando sull’uso dell’Ambion Mix 1 commercialmente disponibile spike-in ad una diluizione di ~2% delle letture finali mappate. Tuttavia, c’è una miscela di dati più vecchi e di dati importati. Pertanto, per tenere traccia degli spike-in utilizzati in una determinata libreria, esiste un set di dati associato alla libreria. Quel dataset conterrà il file di sequenza degli spike-in in formato fasta e le informazioni sulle concentrazioni. Ci si aspetta che queste sequenze spike-in si trovino nell’indice del genoma utilizzato nella fase o nelle fasi di mappatura e nel bam generato successivamente. Le quantificazioni delle sequenze si trovano nei file di trascrizione e quantificazione dei geni RSEM.
Visualizza i dataset spike-in
Visualizza il certificato di analisi per gli spike-in ERCC
Accedi alla dash board ERCC
Links e pubblicazioni
Trova i dati generati da questa pipeline: Tutti | solo paired-end | solo single-end
Esplora le pubblicazioni (in corso)