
- Sequenziamento del genoma Sequencing
- Caratterizzazione genetica
- Metodi di sequenziamento del genoma dell’influenza
Sequenziamento del genoma
I virus dell’influenza cambiano costantemente, infatti tutti i virus influenzali subiscono cambiamenti genetici nel tempo (per maggiori informazioni, vedi Come può cambiare il virus dell’influenza: “Drift” e “Shift”). Il genoma di un virus dell’influenza consiste in tutti i geni che compongono il virus. Il CDC conduce tutto l’anno una sorveglianza dei virus influenzali in circolazione per monitorare i cambiamenti del genoma (o di parti del genoma) di questi virus. Questo lavoro viene eseguito come parte della sorveglianza di routine dell’influenza negli Stati Uniti e come parte del ruolo del CDC come centro di collaborazione dell’Organizzazione Mondiale della Sanità (OMS) per il riferimento e la ricerca sull’influenza. Le informazioni che il CDC raccoglie dallo studio dei cambiamenti genetici (conosciuti anche come “sostituzioni”, “varianti” o “mutazioni”) nei virus dell’influenza giocano un ruolo importante per la salute pubblica aiutando a determinare se i vaccini e i farmaci antivirali funzioneranno contro i virus dell’influenza attualmente in circolazione, così come aiutano a determinare il potenziale dei virus dell’influenza negli animali di infettare gli esseri umani.
Il sequenziamento del genoma rivela la sequenza dei nucleotidi in un gene, come le lettere dell’alfabeto nelle parole. I nucleotidi sono molecole organiche che formano il blocco strutturale degli acidi nucleici, come l’RNA o il DNA. Tutti i virus dell’influenza sono costituiti da RNA a singolo filamento, al contrario del DNA a doppio filamento. I geni RNA dei virus influenzali sono costituiti da catene di nucleotidi legati tra loro e codificati dalle lettere A, C, G e U, che stanno rispettivamente per adenina, citosina, guanina e uracile. Confrontando la composizione dei nucleotidi in un gene del virus con l’ordine dei nucleotidi in un gene di un altro virus si possono rivelare variazioni tra i due virus.
Le variazioni genetiche sono importanti perché possono influenzare la struttura delle proteine di superficie di un virus dell’influenza. Le proteine sono fatte di sequenze di aminoacidi.
La sostituzione di un aminoacido con un altro può influenzare le proprietà di un virus, come ad esempio quanto bene un virus si trasmette tra le persone e quanto il virus è suscettibile ai farmaci antivirali o ai vaccini attuali.
![]()
![]()
Il sequenziamento del genoma rivela la sequenza dei nucleotidi in un gene, come le lettere dell’alfabeto nelle parole. Confrontando la composizione dei nucleotidi di un gene del virus con l’ordine dei nucleotidi di un altro gene del virus, si possono rivelare variazioni tra i due virus.
Le variazioni genetiche sono importanti perché influenzano la struttura delle proteine di superficie del virus dell’influenza. Le proteine sono fatte di sequenze di aminoacidi.
La sostituzione di un aminoacido con un altro può influenzare le proprietà di un virus, come ad esempio quanto bene un virus si trasmette tra le persone e quanto il virus è suscettibile ai farmaci antivirali o ai vaccini attuali.
I virus A e B dell’influenza – i principali virus influenzali che infettano le persone – sono virus RNA che hanno otto segmenti genici. Questi geni contengono “istruzioni” per creare nuovi virus, e sono queste istruzioni che un virus dell’influenza usa una volta che infetta una cellula umana per ingannare la cellula a produrre altri virus dell’influenza, diffondendo così l’infezione.
I geni dell’influenza consistono in una sequenza di molecole chiamate nucleotidi che si legano insieme in una forma a catena. I nucleotidi sono designati dalle lettere A, C, G e U.
Il sequenziamento del genoma è un processo che determina l’ordine, o sequenza, dei nucleotidi (cioè A, C, G e U) in ciascuno dei geni presenti nel genoma del virus. Il sequenziamento del genoma completo può rivelare la sequenza di circa 13.500 lettere di tutti i geni del genoma del virus.
Ogni anno il CDC esegue il sequenziamento del genoma completo su circa 7.000 virus influenzali da campioni clinici originali raccolti attraverso la sorveglianza virologica. Il genoma di un virus A o B dell’influenza contiene otto segmenti genici che codificano (cioè determinano la struttura e le caratteristiche) le 12 proteine del virus, comprese le due proteine di superficie primarie: l’emoagglutinina (HA) e la neuraminidasi (NA). Le proteine di superficie di un virus dell’influenza determinano importanti proprietà del virus, compreso il modo in cui il virus risponde a certi farmaci antivirali, la somiglianza genetica del virus con gli attuali virus vaccinali dell’influenza e il potenziale dei virus influenzali zoonotici (di origine animale) di infettare gli ospiti umani.
Caratterizzazione genetica
Il CDC e altri laboratori di salute pubblica nel mondo hanno sequenziato i geni dei virus dell’influenza fin dagli anni 80. Il CDC contribuisce alle sequenze dei geni alle banche dati pubbliche, come GenBankexternal icon e la Global Initiative on Sharing Avian Influenza Data (GISAID)external icon, per l’uso da parte dei ricercatori di salute pubblica. Le librerie di sequenze genetiche che ne risultano permettono al CDC e ad altri laboratori di confrontare i geni dei virus influenzali attualmente in circolazione con i geni dei vecchi virus influenzali e dei virus usati nei vaccini. Questo processo di confronto delle sequenze genetiche è chiamato caratterizzazione genetica. Il CDC usa la caratterizzazione genetica per le seguenti ragioni:
- Per determinare quanto i virus dell’influenza siano geneticamente “imparentati” o simili tra loro
- Per monitorare come i virus dell’influenza si stanno evolvendo
- Per identificare i cambiamenti genetici che influenzano le proprietà del virus. Per esempio, per identificare i cambiamenti specifici che sono associati ai virus dell’influenza che si diffondono più facilmente, che causano malattie più gravi, o che sviluppano resistenza ai farmaci antivirali
- Per valutare quanto bene un vaccino contro l’influenza potrebbe proteggere contro un particolare virus dell’influenza in base alla sua somiglianza genetica con il virus
- Per monitorare i cambiamenti genetici nei virus dell’influenza che circolano nelle popolazioni animali che potrebbero permettere loro di infettare gli umani.
Le differenze relative tra un gruppo di virus influenzali sono mostrate organizzandole in un grafico chiamato “albero filogenetico”. Gli alberi filogenetici per i virus dell’influenza sono come gli alberi genealogici per le persone. Questi alberi mostrano quanto strettamente ‘imparentati’ siano i singoli virus l’uno con l’altro. I virus sono raggruppati in base al fatto che i nucleotidi dei loro geni siano identici o meno. Gli alberi filogenetici dei virus dell’influenza di solito mostrano quanto sono simili tra loro i geni dell’emoagglutinina (HA) o della neuraminidasi (NA) dei virus. Ogni sequenza di uno specifico virus influenzale ha il proprio ramo sull’albero. Il grado di differenza genetica (numero di differenze nucleotidiche) tra i virus è rappresentato dalla lunghezza delle linee orizzontali (rami) nell’albero filogenetico. Più i virus sono distanti sull’asse orizzontale di un albero filogenetico, più i virus sono geneticamente diversi tra loro.
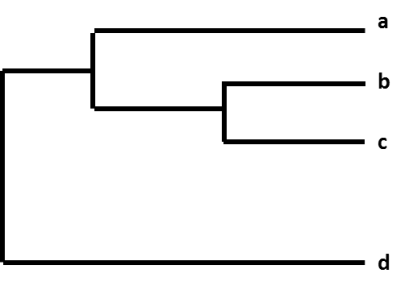
Figura. Un albero filogenetico.
Per esempio, dopo che CDC sequenzia un virus dell’influenza A(H3N2) raccolto attraverso la sorveglianza, la sequenza del virus viene catalogata con altre sequenze di virus che hanno un gene HA simile (H3), e un gene NA simile (N2). Come parte di questo processo, CDC confronta la nuova sequenza del virus con le altre sequenze di virus, e cerca le differenze tra loro. CDC usa poi un albero filogenetico per rappresentare visivamente quanto i virus A(H3N2) siano geneticamente diversi l’uno dall’altro.
CDC esegue la caratterizzazione genetica dei virus influenzali tutto l’anno. Questi dati genetici sono usati insieme ai dati di caratterizzazione antigenica dei virus per aiutare a determinare quali virus vaccinali dovrebbero essere scelti per i prossimi vaccini contro l’influenza dell’emisfero settentrionale o meridionale. Nei mesi che precedono le riunioni di consultazione dell’OMS sui vaccini a febbraio e settembre, il CDC raccoglie i virus influenzali attraverso la sorveglianza e confronta le sequenze dei geni HA e NA degli attuali virus vaccinali con quelle dei virus influenzali in circolazione. Questo è un modo per valutare quanto i virus dell’influenza in circolazione siano strettamente correlati ai virus contro cui è stato formulato il vaccino contro l’influenza stagionale. Man mano che i virus vengono raccolti e caratterizzati geneticamente, le differenze possono essere rivelate.
Per esempio, a volte nel corso di una stagione, i virus circolanti cambiano geneticamente, il che li fa diventare diversi dal corrispondente virus del vaccino. Questa è un’indicazione che potrebbe essere necessario selezionare un virus vaccinale diverso per il vaccino della prossima stagione influenzale, anche se altri fattori, compresi i risultati della caratterizzazione antigenica, influenzano pesantemente le decisioni sul vaccino. Le proteine di superficie HA e NA dei virus influenzali sono antigeni, il che significa che sono riconosciuti dal sistema immunitario e sono in grado di innescare una risposta immunitaria, compresa la produzione di anticorpi che possono bloccare l’infezione. La caratterizzazione antigenica si riferisce all’analisi della reazione di un virus con gli anticorpi per aiutare a valutare come si relaziona con un altro virus.
Metodi di sequenziamento del genoma dell’influenza
Un campione di influenza contiene molte particelle di virus dell’influenza che sono state coltivate in una provetta e che spesso hanno piccole differenze genetiche l’una rispetto all’altra nell’intera popolazione di virus fratelli.
Tradizionalmente, gli scienziati hanno usato una tecnica di sequenziamento chiamata “reazione Sanger” per monitorare l’evoluzione dell’influenza come parte della sorveglianza virologica. Il sequenziamento Sanger identifica la sequenza genetica predominante tra i molti virus influenzali trovati in un isolato. Questo significa che piccole variazioni nella popolazione di virus presenti in un campione non si riflettono nel risultato finale. Gli scienziati spesso usano il metodo Sanger per condurre il sequenziamento parziale del genoma dei virus dell’influenza, mentre le nuove tecnologie (vedi il prossimo paragrafo) sono più adatte al sequenziamento dell’intero genoma.
Negli ultimi cinque anni, il CDC ha utilizzato le metodologie di “Next Generation Sequencing (NGS)”, che hanno notevolmente ampliato la quantità di informazioni e dettagli che l’analisi di sequenziamento può fornire. NGS utilizza il rilevamento molecolare avanzato (AMD) per identificare le sequenze genetiche di ogni virus in un campione. Pertanto, NGS rivela le variazioni genetiche tra molte diverse particelle di virus dell’influenza in un singolo campione, e questi metodi rivelano anche l’intera regione codificante dei genomi. Questo livello di dettaglio può avvantaggiare direttamente il processo decisionale in materia di salute pubblica in modi importanti, ma i dati devono essere attentamente interpretati da esperti altamente addestrati nel contesto di altre informazioni disponibili. Vedi Progetti AMD: Migliorare i vaccini contro l’influenza per ulteriori informazioni su come NGS e AMD stanno rivoluzionando la mappatura del genoma dell’influenza al CDC.
.