
パイプライン概要
Bulk RNA-seq pipelineはENCODE Uniform Processing Pipelinesシリーズの一部として開発されたものである。 パイプラインのフルコードはGithubで自由に利用でき、DNAnexus(リンク先はアカウント作成が必要)で現在の価格で実行することが可能です。
ENCODE Bulk RNA-seq pipelineは、複製と非複製の両方、ペアエンドとシングルエンド、および鎖特異的と非鎖特異的のRNA-seqライブラリに使用することができます。 ライブラリーは、約200 bpより長くなるようにサイズ選択されたmRNA(poly(A)+、rRNA枯渇total RNA、またはpoly(A)-集団)から生成する必要があります。 将来的には、このパイプラインはPAS-seqやBru-seqのデータ処理にも使用できるようになるかもしれない。
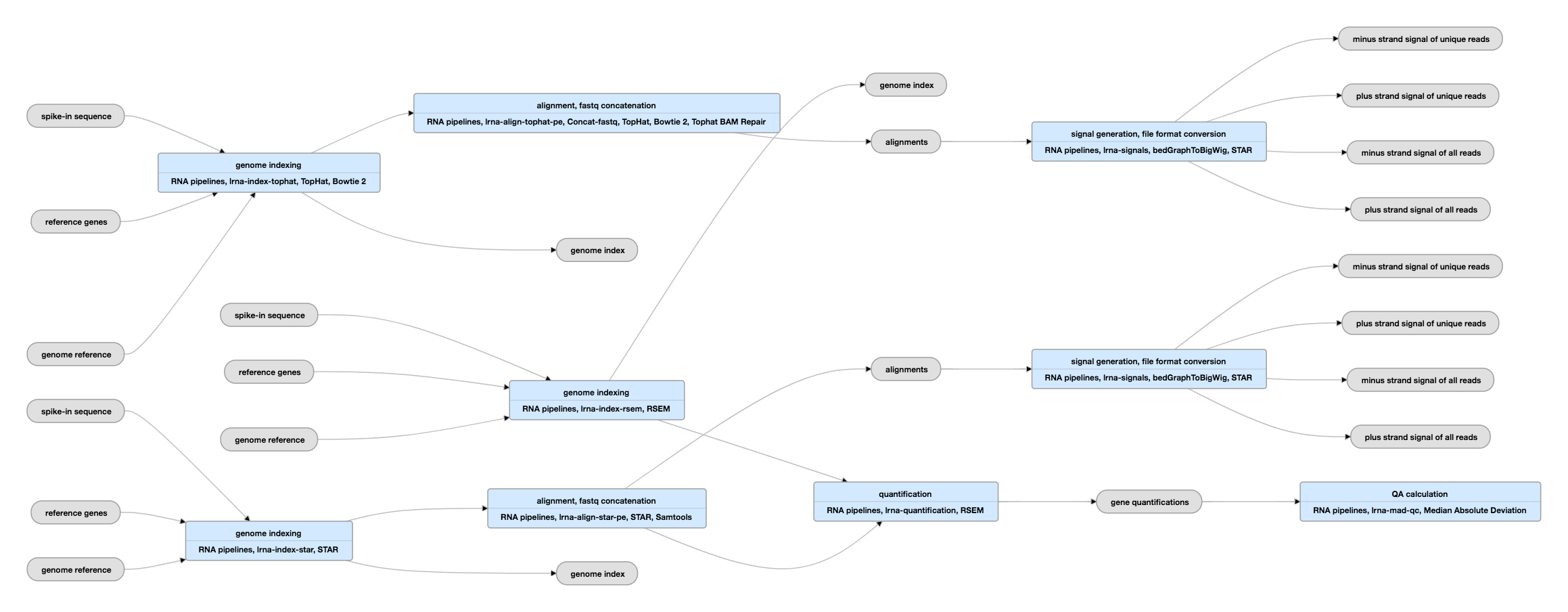
Pipeline Schematic for paired-ended data
View the current instance of this pipeline for paired-ended data
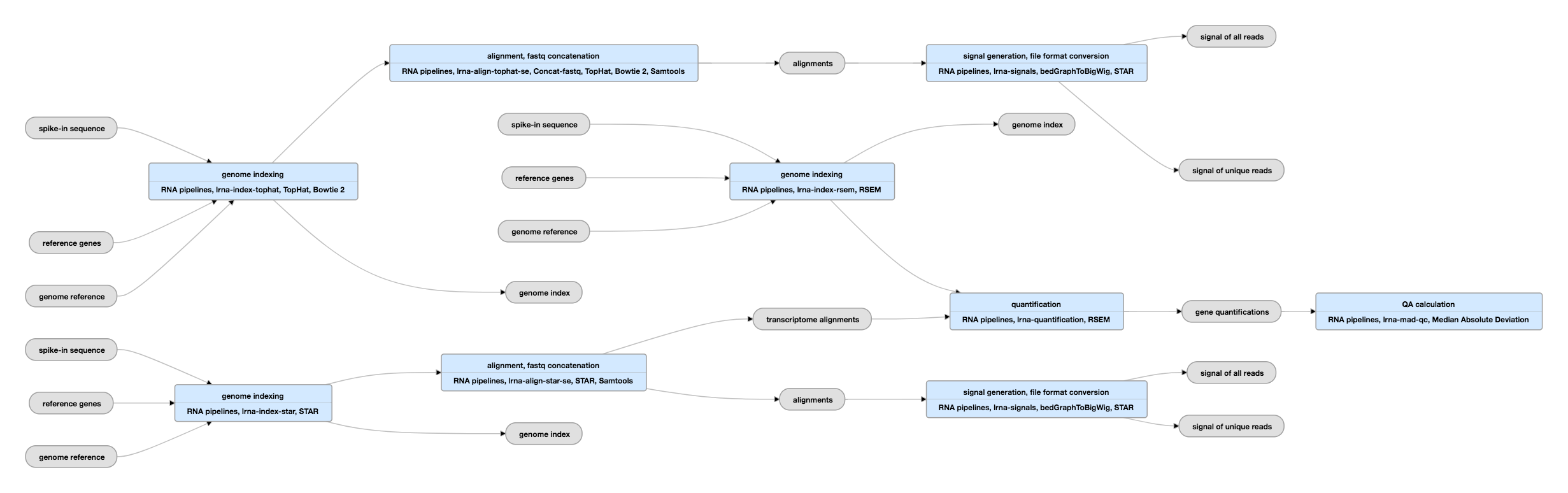
Pipeline Schematic for single-ended data
View the current instances of this pipeline for single-ended data
Inputs.LSI は、以下のようなデータを入力します。
| ファイル形式 |
ファイルに含まれる情報 |
File 説明 |
Note |
| fastq |
reads |
G-…zipped bulk RNA-seq reads | Reads must meet the criteria outlined in the Uniform Processing Pipeline Restrictions. |
| tar | genome index | Generated by STAR or TopHat | alignerとそのインデックスについては、「出力」表下の「アライメントと定量について」パラグラフをご覧ください。 |
| fasta | spike-in sequence | ERCC Spike-ins (External RNA Control Consortium) | スパイクインは事実上RNA-seq実験のコントロールとなるものである。 |
出力される。
| ファイルフォーマット |
ファイルに含まれる情報 |
ファイルの説明 |
Notes |
| bam | alignments | リードをゲノムにマッピングして生成されたもの。 | アライナーとそのインデックスについては、「Outputs」表の下にある「アライメントと定量化について」というパラグラフをご覧ください。 |
| bam | transcriptomeアライメント | 読み取ったデータとトランスクリプトームとの対応付けにより作成します。 | |
| bigWig | signal | 正規化 RNA-seq signal | Strandデータでは、プラス鎖とマイナス鎖の両方でユニークリードとユニーク+マルチマッピングリードについてシグナルを生成しています。 鎖がないデータでは、鎖の同一性に関係なく、ユニークリードとユニーク+マルチマッピングリードに対してシグナルが生成されます。 |
| tsv | gene quantifications | Includes the spike-ins quantifications |
ファイルフォーマットの仕様は、以下の通りです。
olumn 3: length |
| tsv | 転写物定量 | スパイクイン定量 | 下記「アライメントと定量について」項にある「転写物定量に関する注意」を参照ください。 |
| パイプラインは、スピアマン相関やread depthなどの品質メトリクスも生成しています。 | |||
アラインメントと定量化について:
リードのマッピングはSTARプログラム(場合によってはSTARとTopHatアライナーの両方を使用して別々のbamファイルを作成します)、遺伝子や転写物の定量化はRSEMプログラムを使って行われます。 異なるRNA-seqパイプラインによって作成されたマッピングと遺伝子定量には一般的な一致が見られますが、個々の転写産物のアイソフォームの定量はより複雑であり、採用した処理パイプラインによって大幅に異なる場合があり、その精度は未知数です。
Genomic References
View the genome references and chromosome sizes used in this pipeline
The pipelines requires both assembly information for the species of interest and a gene reference. 主なプログラムであるTopHat、STAR、RSEMはそれぞれ、後続のステップで使用するためのインデックスを作成する。 RSEMの使用に関する詳細はこちら
Exogenous RNA Spike-in controls
Exogeneous RNA Spike-in controlsは、RNA発現を定量するための標準ベースラインを作るためにサンプルに加えられる(PMC3166838)。 ENCODEコンソーシアムでは、Ambion Mix 1という市販のスパイクインを、最終的なマッピングリードの2%程度に希釈して使用することを標準化しています。 ただし、古いデータやインポートデータが混在しています。 そのため、あるライブラリで使用されたスパイクインを追跡するために、そのライブラリに関連するデータセットが存在します。 そのデータセットには、スパイクインのシーケンスファイル(Fasta形式)と濃度に関する情報が含まれます。 これらのスパイクイン配列は、マッピングステップで使用されたゲノムインデックスや、その後に生成されたbamに含まれることが予想されます。
スパイクインデータセットを見る
ERCCスパイクインの解析証明書を見る
ERCCダッシュボードにアクセスする
リンクと論文
このパイプラインで生成したデータを検索することが可能です。 すべて|ペアエンドのみ|シングルエンドのみ
出版物の検索(進行中)
。