
- Genome Sequencing
- Genetic Characterization
- Method of Flu Genome Sequencing
Genome Sequencing
インフルエンザウイルスは絶えず変化しています。 実際、すべてのインフルエンザウイルスは時間の経過とともに遺伝子が変化しています(詳しくは、「インフルエンザウイルスはどのように変化するのか」をご覧ください)。 “Drift “と “Shift “をご覧ください)。 インフルエンザウイルスのゲノムは、ウイルスを構成するすべての遺伝子で構成されています。 CDC では、循環するインフルエンザ・ウイルスのゲノム(またはゲノムの一部)の変化を監視するため、年間を通じてサーベイランスを行っています。 この作業は、通常の米国インフルエンザ・サーベイランスの一環として、また世界保健機関(WHO)のインフルエンザに関する調査・研究のための協力センターとしてのCDCの役割の一環として行われています。 CDCがインフルエンザウイルスの遺伝的変化(「置換」、「変異」、「突然変異」としても知られています)を研究して収集した情報は、ワクチンや抗ウイルス剤が現在流通しているインフルエンザウイルスに対して有効かどうかを判断したり、動物のインフルエンザウイルスが人に感染する可能性を判断するのに役立つなど、公衆衛生上重要な役割を担っています。 ヌクレオチドは、RNAやDNAなどの核酸の構造単位ビルディングブロックを形成する有機分子である。 インフルエンザウイルスはすべて一本鎖のRNAからなり、二本鎖のDNAとは対照的である。 インフルエンザウイルスのRNA遺伝子は、ヌクレオチドが鎖状に結合したもので、アデニン、シトシン、グアニン、ウラシルをそれぞれ表すA、C、G、Uという文字でコードされている。 あるウイルス遺伝子のヌクレオチドの構成と、別のウイルス遺伝子のヌクレオチドの順番を比較することで、2つのウイルス間の変異を明らかにすることができます。
遺伝子の変異は、インフルエンザウイルスの表面タンパク質の構造に影響を与えることができるので重要です。 タンパク質はアミノ酸の配列でできています。
あるアミノ酸を別のアミノ酸に置き換えることで、ウイルスが人々の間でどの程度感染するか、抗ウイルス薬や現在のワクチンに対してどの程度感受性があるかなど、ウイルスの性質に影響を与えることができます。 CDCは次世代シーケンシングとタンパク質によってインフルエンザワクチンを改善します。 詳細はリンクをご覧ください。
![]()
ゲノム配列決定により、言葉のアルファベットのように、遺伝子内のヌクレオチドの配列が明らかになりました。 あるウイルス遺伝子のヌクレオチドの構成と、別のウイルス遺伝子のヌクレオチドの順序を比較することで、2つのウイルス間の変異を明らかにすることができます。
遺伝子の変異は、インフルエンザウイルスの表面タンパク質の構造に影響を与えるため重要です。 タンパク質はアミノ酸の配列でできています。
あるアミノ酸を別のアミノ酸に置き換えることで、ウイルスが人々の間でどのくらい感染するか、抗ウイルス薬や現在のワクチンに対してどのくらい感受性があるかなど、ウイルスの性質に影響を与えることができます。 これらの遺伝子には、新しいウイルスを作るための「命令」が含まれており、インフルエンザウイルスがヒトの細胞に感染すると、この命令を使って、細胞を騙してさらにインフルエンザウイルスを作らせ、感染を拡大させるのです。 ゲノム配列決定とは、ウイルスのゲノムに存在する各遺伝子におけるヌクレオチド(すなわちA、C、G、U)の順番、または配列を決定するプロセスです。 全ゲノム配列決定により、ウイルスゲノムの全遺伝子の約13,500文字の配列が明らかになります。
毎年CDCは、ウイルス学的サーベイランスを通じて収集したオリジナルの臨床サンプルから約7000のインフルエンザウイルスの全ゲノム配列決定を行っています。 A型またはB型インフルエンザウイルスのゲノムには、ヘマグルチニン(HA)とノイラミニダーゼ(NA)という2つの主要な表面タンパク質を含む、ウイルスの12種類のタンパク質をコードする(すなわち、その構造と特徴を決定する)8つの遺伝子断片が含まれています。 インフルエンザウイルスの表面タンパク質は、特定の抗ウイルス剤に対するウイルスの反応性、現在のインフルエンザワクチンウイルスとの遺伝的類似性、人獣共通感染症(動物由来)インフルエンザウイルスがヒト宿主に感染する可能性などの、ウイルスの重要な特性を決定しています。 CDCは、公衆衛生研究者が利用できるように、遺伝子配列をGenBanke(外部サイト)やGISAID(外部サイト)などの公的データベースに提供しています。 こうして得られた遺伝子配列のライブラリにより、CDCやその他の研究所は、現在流通しているインフルエンザウイルスの遺伝子と、古いインフルエンザウイルスやワクチンに使用されるウイルスの遺伝子を比較することができます。 この遺伝子配列を比較するプロセスは、遺伝子特性評価と呼ばれています。 CDCは以下の理由で遺伝子解析を行います:
- インフルエンザウイルスが遺伝子的にどれだけ「近縁」または類似しているかを判断するため
- インフルエンザウイルスがどのように進化しているかを監視するため
- ウイルスの特性に影響を及ぼす遺伝的変化を特定するため。 例えば、インフルエンザウイルスがより容易に広がること、より重篤な疾患を引き起こすこと、または抗ウイルス剤に対する耐性を発達させることに関連する特定の変化を特定するため<5134><1835>ウイルスとの遺伝的類似性に基づいて、インフルエンザインフルエンザワクチンが特定のインフルエンザウイルスに対してどの程度の予防効果があるかを評価するため<5134><1835>動物集団に循環するインフルエンザウイルスにヒトへの感染が可能となる遺伝的変化がないか監視するため。
インフルエンザウイルスのグループ間の相対的な違いは、「系統樹」と呼ばれる図形に整理することで示されます。 インフルエンザウイルスの系統樹は、人間でいえば家系(家系図)のようなものです。 これらの樹は、個々のウイルスが互いにどれだけ「近縁」であるかを示しています。 ウイルスは、遺伝子のヌクレオチドが同じかどうかでグループ分けされる。 インフルエンザウイルスの系統樹は通常、ウイルスのヘマグルチニン(HA)またはノイラミニダーゼ(NA)遺伝子が互いにどれだけ似ているかを表示します。 特定のインフルエンザウイルスからの各配列は、樹上のそれぞれの枝を持ちます。 ウイルス間の遺伝的な違いの度合い(ヌクレオチドの違いの数)は、系統樹の横線(枝)の長さで表されます。 系統樹の横軸上でウイルスが離れているほど、ウイルスの遺伝的な違いが大きいことを意味する
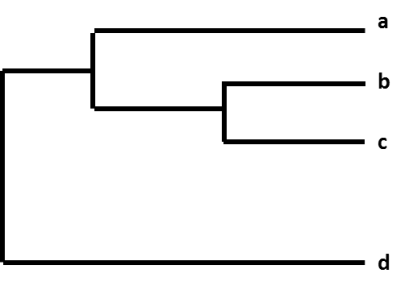
図. 系統樹。
例えば、CDCがサーベイランスで収集したインフルエンザA(H3N2)ウイルスの配列を決定した後、そのウイルス配列を、同様のHA遺伝子(H3)および同様のNA遺伝子(N2)を有する他のウイルス配列とカタログ化する。 このプロセスの一環として、CDCは新しいウイルス配列を他のウイルス配列と比較し、それらの間の相違点を探します。 CDC はその後、系統樹を使用して、A(H3N2)ウイルスが互いにどの程度遺伝的に異なるかを視覚的に表現します。
CDC は年間を通じてインフルエンザウイルスの遺伝子特性解析を行っています。 この遺伝子データは、ウイルスの抗原特性データと合わせて、今後の北半球または南半球のインフルエンザ・ワクチンにどのワクチン・ウイルスを選択すべきかを決定するために使用されます。 2月と9月のWHOワクチン協議会までの数ヶ月間、CDCはサーベイランスを通じてインフルエンザウイルスを収集し、現在のワクチンウイルスのHAおよびNA遺伝子配列を、流通しているインフルエンザウイルスのものと比較しています。 これは、循環しているインフルエンザウイルスが、季節性インフルエンザワクチンを予防するために処方されたウイルスとどれだけ密接に関連しているかを評価する一つの方法です。 例えば、あるシーズンの間に、循環しているウイルスが遺伝的に変化し、対応するワクチンウイルスと異なるものになることがあります。 これは、次のシーズンのワクチンとして異なるワクチンウイルスを選択する必要があることを示す1つの指標ですが、抗原特性に関する知見を含む他の要因も、ワクチンの決定に大きく影響します。 インフルエンザウイルスのHAおよびNAの表面タンパク質は抗原であり、免疫系によって認識され、感染を阻止する抗体の産生などの免疫反応を引き起こすことができる。 4646>
インフルエンザゲノム配列解析の方法
1つのインフルエンザサンプルには、試験管内で増殖した多数のインフルエンザウイルス粒子が含まれており、兄弟ウイルス集団全体の中で互いに比較して遺伝子の違いが小さいことがよくあります。
従来、科学者は、ウイルス学的サーベイランスの一環として、「サンガー反応」と呼ばれる配列決定技術を使って、インフルエンザの進化を監視してきました。 サンガー配列決定では、分離された多くのインフルエンザウイルスの中から、優勢な遺伝子配列を特定します。 つまり、サンプルに存在するウイルス集団の小さな変動は最終結果に反映されないということです。 科学者はしばしばサンガー法を使用してインフルエンザウイルスの部分ゲノムシーケンスを行いますが、新しいテクノロジー(次項参照)は全ゲノムシーケンスに適しています。
過去 5 年間、CDC は「次世代シーケンス(NGS)」手法を使用しており、シーケンス分析で得られる情報量と詳細が大幅に拡大されています。 NGSは、高度な分子検出(AMD)を用いて、サンプル中の各ウイルスから遺伝子配列を特定します。 そのため、NGSは1つのサンプルに含まれる多くの異なるインフルエンザウイルス粒子間の遺伝的変異を明らかにし、これらの方法はゲノムのコーディング領域全体も明らかにします。 このレベルの詳細は、公衆衛生の意思決定に重要な形で直接役立つ可能性がありますが、データは、他の利用可能な情報との関連において、高度な訓練を受けた専門家が慎重に解釈する必要があります。 AMD プロジェクトをご覧ください。 CDCでNGSとAMDがどのようにインフルエンザゲノムマッピングに革命を起こしているかについての詳細は、AMDプロジェクト:インフルエンザワクチンの改善
を参照してください。