
Pijplijn overzicht
De Bulk RNA-seq pijplijn is ontwikkeld als onderdeel van de ENCODE Uniform Processing Pipelines serie. De volledige pijplijn code is vrij beschikbaar op Github en kan worden uitgevoerd op DNAnexus (link vereist account aanmaken) tegen hun huidige prijzen.
De ENCODE Bulk RNA-seq pijplijn kan worden gebruikt voor zowel gerepliceerde als niet-gerepliceerde, gepaarde of enkelvoudige, en strengspecifieke of niet-strengspecifieke RNA-seq bibliotheken. Bibliotheken moeten worden gegenereerd uit mRNA (poly (A) +, rRNA-verarmd totaal RNA, of poly (A) – populaties die zijn grootte-selected langer te zijn dan ongeveer 200 bp. In de toekomst kan deze pijplijn ook worden gebruikt om PAS-seq en Bru-seq gegevens te verwerken.
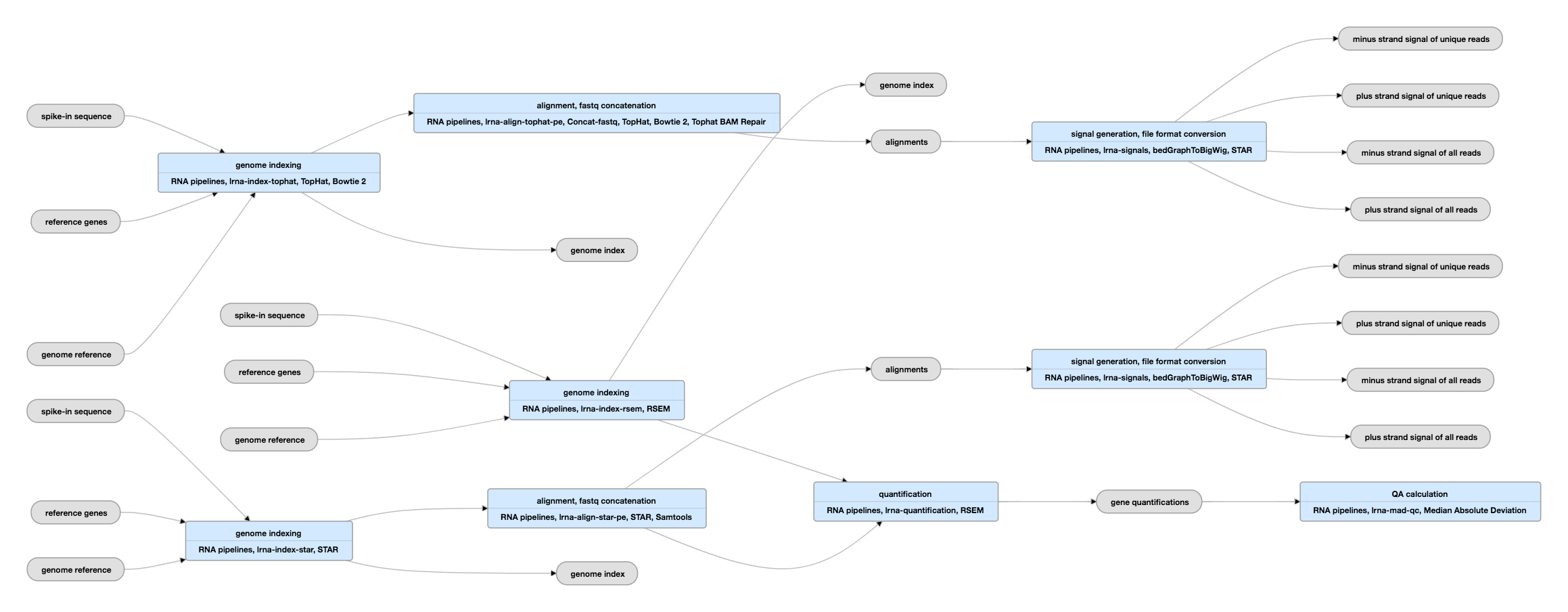
Pijplijn Schema voor gepaarde-ended data
Bekijk de huidige instantie van deze pijplijn voor gepaarde-ended data
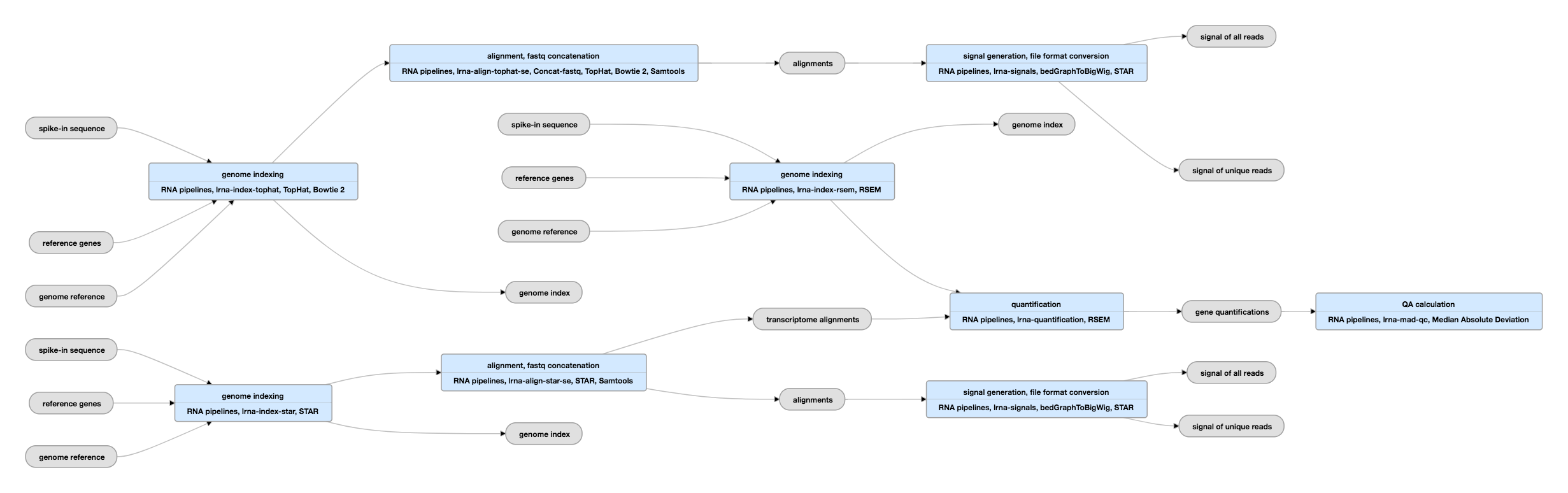
Pijplijn Schema voor single-ended data
Bekijk de huidige instanties van deze pijplijn voor single-ended data
Inputs:
| Bestandsindeling |
Informatie in bestand |
Bestands beschrijving |
Notities |
| fastq |
reads |
G-gezipte bulk RNA-seq reads | Reads moeten voldoen aan de criteria zoals uiteengezet in de Uniform Processing Pipeline Restrictions. |
| tar | genoomindex | Gegenereerd door STAR of TopHat | Zie de paragraaf “Betreffende uitlijning en kwantificering” onder de tabel “Outputs” voor meer informatie over de uitlijners en hun indexen. |
| fasta | spike-in sequence | ERCC Spike-ins (External RNA Control Consortium) | De spike-ins zijn in feite de controles voor het RNA-seq experiment. |
Outputs:
| Bestandsindeling |
Informatie in bestand |
Bestandsbeschrijving |
Notities |
| bam | alignementen | Gemaakt door reads aan het genoom te koppelen. | Zie de paragraaf getiteld “Betreffende alignment en kwantificering” onder de tabel “Outputs” voor meer informatie over de aligners en hun indices. |
| bam | transcriptoom alignments | Geproduceerd door het in kaart brengen van de gelezen naar het transcriptoom. | |
| bigWig | signal | Genormaliseerd RNA-seq signaal | Voor gestrande data worden signalen gegenereerd voor unieke gelezen en unieke+multimapping gelezen in zowel de plus- als de min-streng. Voor unstranded gegevens, worden signalen gegenereerd voor unieke leest en unieke + multimapping leest zonder inachtneming van streng identiteit. |
| tsv | genkwantificaties | Inclusief de spike-ins kwantificaties |
De specificaties van het bestandsformaat zijn als volgt:
|
| tsv | transcriptkwantificaties | Inclusief de spike-ins kwantificaties | Zie de waarschuwing betreffende transcriptkwantificaties in de onderstaande paragraaf met de titel “Betreffende uitlijning en kwantificering”. |
| De pijplijn produceert ook kwaliteitsmetriek, waaronder Spearman-correlatie en leesdiepte. | |||
Met betrekking tot uitlijning en kwantificering:
Het in kaart brengen van de gelezen data gebeurt met behulp van het STAR-programma (in sommige gevallen worden zowel STAR als TopHat aligners gebruikt om afzonderlijke bam-bestanden te produceren) en de kwantificering van genen en transcripten gebeurt met het RSEM-programma. Hoewel er algemene overeenstemming tussen de mappings en het gen kwantificaties die door verschillende RNA-seq pijplijnen, kwantificaties van individuele transcript isovormen, die veel complexer, kan sterk verschillen, afhankelijk van de verwerking pijplijn gebruikt en zijn van onbekende nauwkeurigheid. Daarom uitlijningen en gen kwantificaties kunnen vol vertrouwen worden gebruikt, terwijl transcript kwantificaties moeten worden gebruikt met zorg.
Genomic References
Bekijk het genoom referenties en chromosoom maten gebruikt in deze pijplijn
Deze pijplijnen vereisen zowel de assemblage informatie voor de soort van belang en een gen referentie. Elk van de hoofdprogramma’s, TopHat, STAR en RSEM, maakt een index voor gebruik in de volgende stappen. Meer informatie over het gebruik van RSEM is hier beschikbaar.
Exogene RNA spike-in controles
Exogene RNA spike-in controles worden toegevoegd aan monsters tot een standaard basislijn voor de kwantificering van RNA expressie (PMC3166838) te creëren. De ENCODE consortium is standaardiseren op het gebruik van de Ambion Mix 1 commercieel beschikbare spike-ins bij een verdunning van ~ 2% van de uiteindelijke in kaart gebrachte leest. Er is echter een mengsel van oudere gegevens en geïmporteerde gegevens. Daarom, om de spike-ins gebruikt in een bepaalde bibliotheek bij te houden, is er een dataset geassocieerd met de bibliotheek. Die dataset bevat het sequentiebestand van de spike-ins in fasta-formaat en informatie over de concentraties. Deze spike-in sequenties worden verwacht te vinden in het genoom index gebruikt in de mapping stap (s) en in de vervolgens gegenereerde bam. De kwantificaties van de sequenties zijn te vinden in de RSEM transcript en gen kwantificatie bestanden.
Bekijk spike-ins datasets
Bekijk het analysecertificaat voor ERCC spike-ins
Toegang tot het ERCC dash board
Links en Publicaties
Vind data gegenereerd door deze pijplijn: All | paired-end only | single-end only
Verken publicaties (in uitvoering)