
- Genome Sequencing
- Genetic Characterization
- Methods of Flu Genome Sequencing
Genome Sequencing
Influenzavirussen zijn voortdurend in beweging, in feite ondergaan alle griepvirussen in de loop der tijd genetische veranderingen (voor meer informatie, zie Hoe het griepvirus kan veranderen: “Drift’ en ‘Shift’). Het genoom van een influenzavirus bestaat uit alle genen waaruit het virus is opgebouwd. CDC houdt het hele jaar door toezicht op circulerende influenzavirussen om veranderingen in het genoom (of delen van het genoom) van deze virussen in de gaten te houden. Deze werkzaamheden worden uitgevoerd als onderdeel van de routinesurveillance van de Amerikaanse griep en als onderdeel van de rol van het CDC als Samenwerkend centrum van de Wereldgezondheidsorganisatie (WHO) voor referentie en onderzoek naar griep. De informatie die CDC verzamelt door genetische veranderingen (ook bekend als “substituties”, “varianten” of “mutaties”) in influenzavirussen te bestuderen, speelt een belangrijke rol voor de volksgezondheid doordat het helpt te bepalen of vaccins en antivirale geneesmiddelen zullen werken tegen de huidige circulerende influenzavirussen, en ook helpt te bepalen in hoeverre influenzavirussen in dieren mensen kunnen besmetten.
De sequentie van het genoom onthult de volgorde van de nucleotiden in een gen, zoals alfabetletters in woorden. Nucleotiden zijn organische moleculen die de structurele bouwstenen vormen van nucleïnezuren, zoals RNA of DNA. Alle influenzavirussen bestaan uit enkelstrengs RNA in tegenstelling tot dubbelstrengs DNA. De RNA-genen van influenzavirussen bestaan uit ketens van nucleotiden die aan elkaar zijn gebonden en worden gecodeerd door de letters A, C, G en U, die respectievelijk staan voor adenine, cytosine, guanine en uracil. Door de samenstelling van nucleotiden in een virusgen te vergelijken met de volgorde van nucleotiden in een ander virusgen, kunnen variaties tussen de twee virussen aan het licht komen.
Genetische variaties zijn belangrijk omdat ze de structuur van de oppervlakte-eiwitten van een influenzavirus kunnen beïnvloeden. Eiwitten zijn opgebouwd uit reeksen aminozuren.
De vervanging van het ene aminozuur door het andere kan van invloed zijn op de eigenschappen van een virus, zoals hoe goed een virus tussen mensen overdraagbaar is en hoe vatbaar het virus is voor antivirale geneesmiddelen of de huidige vaccins.
![]()
![]()
Genoomsequencing onthult de volgorde van de nucleotiden in een gen, zoals alfabetletters in woorden. Door de samenstelling van de nucleotiden in een virusgen te vergelijken met de volgorde van de nucleotiden in een ander virusgen kunnen variaties tussen de twee virussen aan het licht worden gebracht.
Genetische variaties zijn belangrijk omdat zij van invloed zijn op de structuur van de oppervlakte-eiwitten van een influenzavirus. Eiwitten zijn opgebouwd uit reeksen aminozuren.
De vervanging van het ene aminozuur door het andere kan van invloed zijn op de eigenschappen van een virus, zoals hoe goed een virus tussen mensen overdraagbaar is, en hoe vatbaar het virus is voor antivirale geneesmiddelen of de huidige vaccins.
Influenza A- en B-virussen – de primaire influenzavirussen die mensen infecteren – zijn RNA-virussen die acht gensegmenten hebben. Deze genen bevatten “instructies” voor het maken van nieuwe virussen, en het zijn deze instructies die een influenzavirus gebruikt wanneer het een menselijke cel infecteert om de cel ertoe te brengen meer influenzavirussen te produceren, waardoor de infectie zich verspreidt.
Influenzagenen bestaan uit een reeks moleculen, nucleotiden genaamd, die in een kettingachtige vorm aan elkaar zijn gehecht. Nucleotiden worden aangeduid met de letters A, C, G en U.
Genome sequencing is een proces dat de volgorde, of sequentie, bepaalt van de nucleotiden (d.w.z. A, C, G en U) in elk van de genen die aanwezig zijn in het genoom van het virus. Full genome sequencing kan de ongeveer 13.500 letters tellende sequentie van alle genen van het genoom van het virus onthullen.
Elk jaar voert het CDC whole genome sequencing uit op ongeveer 7.000 influenzavirussen van originele klinische monsters die via virologische surveillance zijn verzameld. Het genoom van een influenza A- of B-virus bevat acht gensegmenten die coderen voor (d.w.z. de structuur en kenmerken bepalen van) de 12 eiwitten van het virus, waaronder de twee primaire oppervlakte-eiwitten: hemagglutinine (HA) en neuraminidase (NA). De oppervlakte-eiwitten van een influenzavirus bepalen belangrijke eigenschappen van het virus, zoals hoe het virus op bepaalde antivirale geneesmiddelen reageert, de genetische gelijkenis van het virus met de huidige influenzavirusvaccins en de mogelijkheid dat zoönotische influenzavirussen (van dierlijke oorsprong) menselijke gastheren infecteren.
Genetische karakterisering
Het CDC en andere volksgezondheidslaboratoria over de hele wereld zijn al sinds de jaren tachtig bezig met het sequencen van de genen van influenzavirussen. CDC draagt gensequenties bij aan openbare databanken, zoals GenBankextern icoon en het Global Initiative on Sharing Avian Influenza Data (GISAID)extern icoon, voor gebruik door onderzoekers op het gebied van de volksgezondheid. De resulterende bibliotheken van gensequenties stellen CDC en andere laboratoria in staat de genen van momenteel circulerende influenzavirussen te vergelijken met de genen van oudere influenzavirussen en virussen die in vaccins worden gebruikt. Dit proces van het vergelijken van genetische sequenties wordt genetische karakterisering genoemd. CDC gebruikt genetische karakterisering om de volgende redenen:
- Om te bepalen hoe nauw “verwant” of vergelijkbaar griepvirussen genetisch met elkaar zijn
- Om te volgen hoe griepvirussen evolueren
- Om genetische veranderingen te identificeren die de eigenschappen van het virus beïnvloeden. Bijvoorbeeld om de specifieke veranderingen te identificeren die worden geassocieerd met griepvirussen die zich gemakkelijker verspreiden, meer ernstige ziekte veroorzaken of resistentie tegen antivirale geneesmiddelen ontwikkelen
- Om te beoordelen hoe goed een griepvaccin tegen een bepaald griepvirus zou kunnen beschermen op basis van de genetische gelijkenis met het virus
- Om te controleren op genetische veranderingen in griepvirussen die in dierpopulaties circuleren en die hen in staat zouden kunnen stellen mensen te infecteren.
De relatieve verschillen tussen een groep influenzavirussen worden getoond door ze te ordenen in een grafiek die een ‘fylogenetische boom’ wordt genoemd. Fylogenetische bomen voor influenzavirussen zijn als familie (genealogie) bomen voor mensen. Deze bomen laten zien hoe “verwant” individuele virussen aan elkaar zijn. Virussen worden gegroepeerd op basis van het feit of de nucleotiden van hun genen identiek zijn of niet. Fylogenetische bomen van influenzavirussen laten meestal zien hoe sterk de hemagglutinine- (HA) of neuraminidase- (NA) genen van de virussen op elkaar lijken. Elke sequentie van een specifiek influenzavirus heeft zijn eigen tak in de boom. De mate van genetisch verschil (aantal nucleotideverschillen) tussen virussen wordt weergegeven door de lengte van de horizontale lijnen (takken) in de fylogenetische boom. Hoe verder de virussen van elkaar verwijderd zijn op de horizontale as van een fylogenetische boom, hoe meer de virussen genetisch van elkaar verschillen.
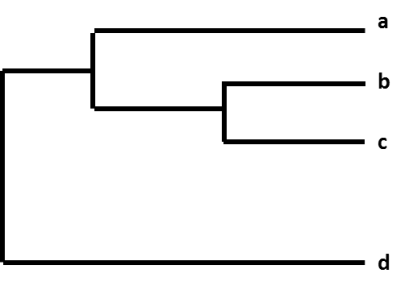
Figuur. Een fylogenetische boom.
Als CDC bijvoorbeeld een sequentie heeft vastgesteld van een influenza A(H3N2)-virus dat via surveillance is verzameld, wordt de virussequentie gecatalogiseerd met andere virussequenties die een vergelijkbaar HA-gen (H3) en een vergelijkbaar NA-gen (N2) hebben. Als onderdeel van dit proces vergelijkt het CDC de nieuwe virussequentie met de andere virussequenties, en zoekt het naar onderlinge verschillen. CDC maakt vervolgens gebruik van een fylogenetische boom om visueel weer te geven hoe genetisch de A(H3N2)-virussen van elkaar verschillen.
CDC voert het hele jaar door genetische karakterisering van influenzavirussen uit. Deze genetische gegevens worden gebruikt in combinatie met gegevens over virusantigene karakterisering om te helpen bepalen welke vaccinevirussen moeten worden gekozen voor de komende griepvaccins voor het noordelijk halfrond of het zuidelijk halfrond. In de maanden voorafgaand aan de WHO-vergaderingen over vaccins in februari en september verzamelt het CDC influenzavirussen via surveillance en vergelijkt het de HA- en NA-gensequenties van de huidige vaccinevirussen met die van circulerende griepvirussen. Dit is één manier om na te gaan hoe nauw de circulerende influenzavirussen verwant zijn met de virussen waartegen het seizoensgriepvaccin bescherming moest bieden. Wanneer virussen worden verzameld en genetisch worden gekarakteriseerd, kunnen verschillen aan het licht komen.
Zo zullen circulerende virussen soms in de loop van een seizoen genetisch veranderen, waardoor zij gaan verschillen van het corresponderende vaccinvirus. Dit is één aanwijzing dat voor het vaccin van het volgende griepseizoen misschien een ander vaccinvirus moet worden geselecteerd, hoewel andere factoren, waaronder bevindingen op het gebied van antigene karakterisering, van grote invloed zijn op beslissingen over vaccins. De HA- en NA-oppervlakte-eiwitten van influenzavirussen zijn antigenen, wat betekent dat ze door het immuunsysteem worden herkend en in staat zijn een immuunrespons op te wekken, waaronder de productie van antilichamen die de infectie kunnen blokkeren. Antigenische karakterisering verwijst naar de analyse van de reactie van een virus met antilichamen om te helpen beoordelen hoe het zich verhoudt tot een ander virus.
Methoden voor sequentiebepaling van het genoom van griep
Een influenzamonster bevat veel influenzavirusdeeltjes die in een reageerbuis zijn gekweekt en die vaak kleine genetische verschillen vertonen in vergelijking met elkaar in de hele populatie van broervirussen.
Traditioneel maken wetenschappers gebruik van een sequencingtechniek die “de Sanger-reactie” wordt genoemd om de evolutie van influenza te volgen als onderdeel van virologisch toezicht. Sanger-sequencing identificeert de overheersende genetische sequentie onder de vele influenzavirussen die in een isolaat worden aangetroffen. Dit betekent dat kleine variaties in de populatie van virussen in een monster niet in het eindresultaat tot uiting komen. Wetenschappers gebruiken de Sanger-methode vaak om gedeeltelijke genoomsequencing van influenzavirussen uit te voeren, terwijl nieuwere technologieën (zie volgende alinea) beter geschikt zijn voor sequencing van het volledige genoom.
De afgelopen vijf jaar heeft het CDC gebruik gemaakt van “Next Generation Sequencing (NGS)”-methodologieën, die de hoeveelheid informatie en details die sequencinganalyse kan opleveren, sterk hebben uitgebreid. NGS maakt gebruik van geavanceerde moleculaire detectie (AMD) om gensequenties van elk virus in een monster te identificeren. NGS brengt dus de genetische variaties tussen vele verschillende influenzavirusdeeltjes in één monster aan het licht, en deze methoden brengen ook de volledige coderende regio van de genomen aan het licht. Dit niveau van detail kan de besluitvorming op het gebied van de volksgezondheid in belangrijke mate ten goede komen, maar de gegevens moeten door hoogopgeleide deskundigen zorgvuldig worden geïnterpreteerd in de context van andere beschikbare informatie. Zie AMD-projecten: Improving Influenza Vaccines voor meer informatie over hoe NGS en AMD een revolutie teweegbrengen in het in kaart brengen van het griepgenoom bij CDC.