
Przegląd linii
Potok Bulk RNA-seq został opracowany jako część serii ENCODE Uniform Processing Pipelines. Pełny kod potoku jest swobodnie dostępny na Github i może być uruchomiony na DNAnexus (link wymaga utworzenia konta) po ich aktualnych cenach.
Potok ENCODE Bulk RNA-seq może być używany zarówno dla replikowanych jak i niereplikowanych, paired-ended lub single-ended, oraz specyficznych lub nie specyficznych dla danego pasma bibliotek RNA-seq. Biblioteki muszą być generowane z mRNA (poli(A)+, całkowite RNA pozbawione rRNA, lub populacje poli(A)-, które są selekcjonowane pod względem wielkości, aby były dłuższe niż około 200 bp. W przyszłości ten potok może być również używany do przetwarzania danych PAS-seq i Bru-seq.
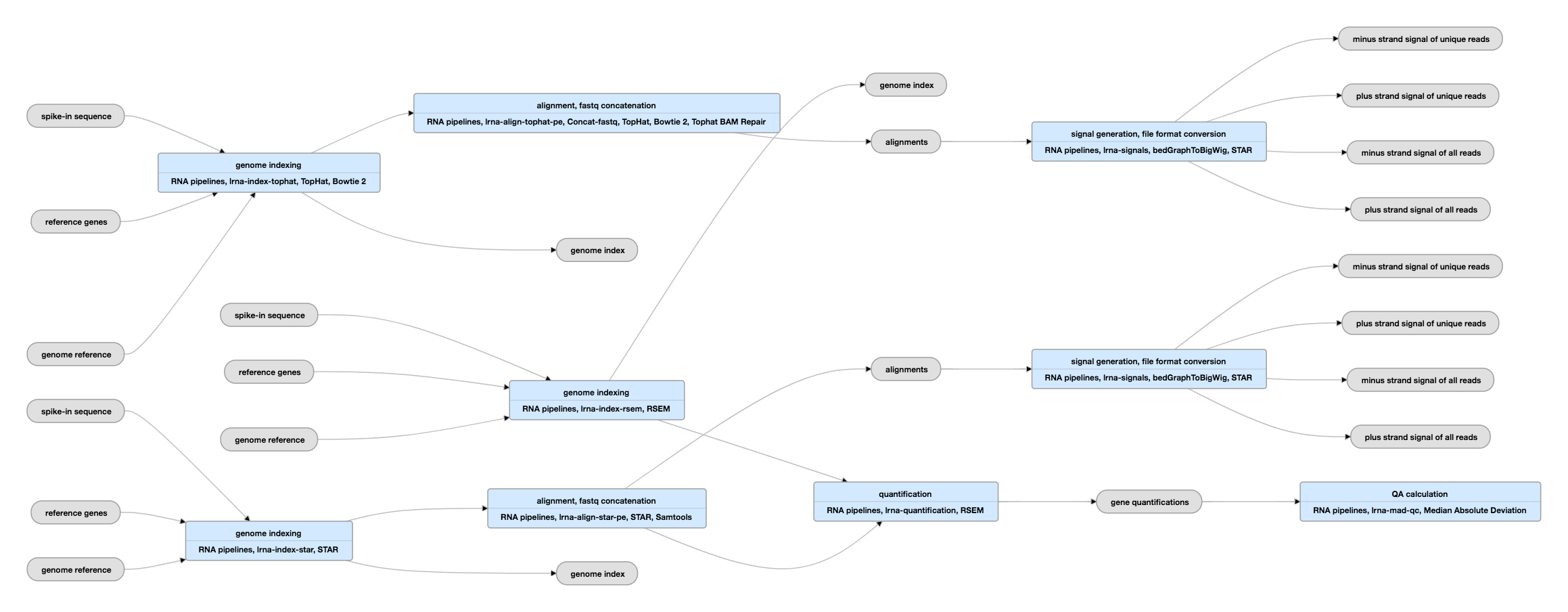
Schemat rurociągu dla danych typu paired-ended
Zobacz bieżącą instancję tego rurociągu dla danych typu paired-ended
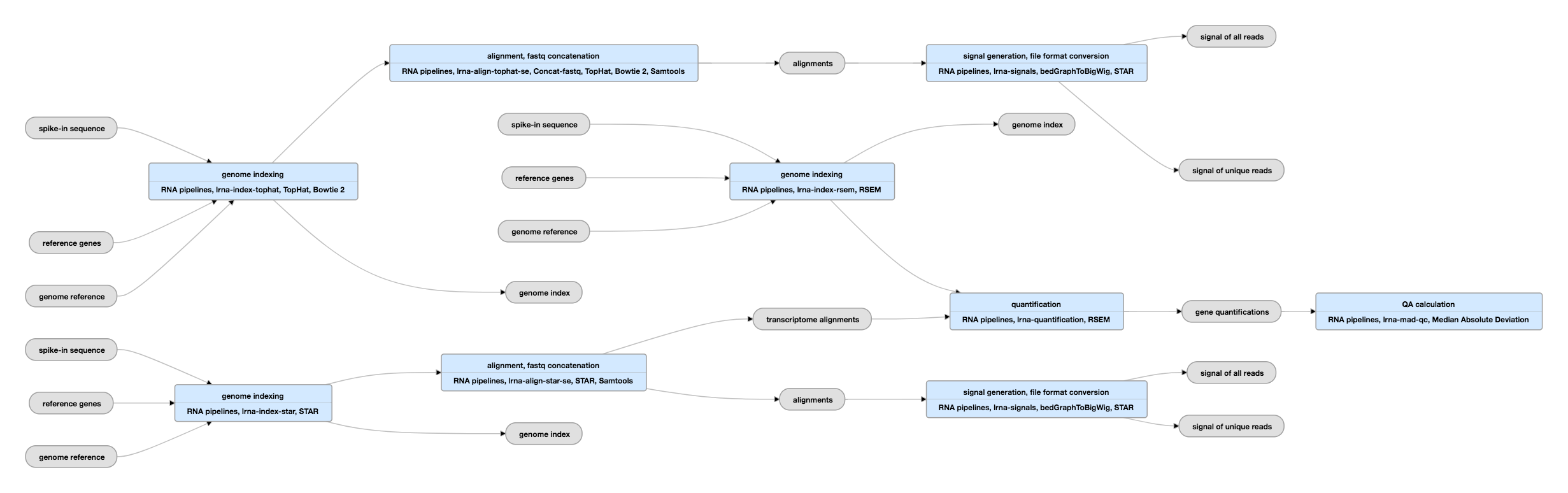
Schemat rurociągu dla danych typu single-ended
Zobacz bieżącą instancję tego rurociągu dla danych typu single-ended
Wejścia:
| Format pliku |
Informacje zawarte w pliku |
Plik. description |
Notes |
| fastq |
reads |
G-.zripowane odczyty RNA-seq luzem | Odczyty muszą spełniać kryteria przedstawione w Ograniczeniach Jednolitego Potoku Przetwarzania. |
| tar | genome index | Generated by STAR or TopHat | Proszę zobaczyć akapit zatytułowany „Regarding alignment and quantification” poniżej tabeli „Outputs”, aby dowiedzieć się więcej o alignerach i ich indeksach. |
| fasta | sekwencja spike-in | Sekwencje spike-in ERCC (External RNA Control Consortium) | Sekwencje spike-in są efektywnie kontrolami dla eksperymentu RNA-seq. |
Wyniki:
| Format pliku |
Informacje zawarte w pliku |
Opis pliku |
Notes |
| bam | alignments | Produced by mapping reads to the genome. | Zapoznaj się z akapitem zatytułowanym „Regarding alignment and quantification” pod tabelą „Outputs”, aby dowiedzieć się więcej o alignerach i ich indeksach. |
| bam | transcriptome alignments | Produced by mapping reads to the transcriptome. | |
| bigWig | signal | Normalizowany sygnał RNA-seq | Dla danych splotowych, sygnały są generowane dla unikalnych odczytów i unikalnych+multimappingowych odczytów w obu splotach plus i minus. Dla danych nie splotowych, sygnały są generowane dla unikalnych odczytów i unikalnych+multimapujących odczytów bez względu na tożsamość splotu. |
| tsv | kwantyfikacje genów | Zawiera kwantyfikacje spike-ins |
Specyfikacje formatu pliku są następujące:
|
| tsv | kwantyfikacje transkryptów | Zawiera kwantyfikacje spike-ins | Proszę zapoznać się z przestrogą dotyczącą kwantyfikacji transkryptów w akapicie poniżej zatytułowanym „Regarding alignment and quantification”. |
| Potok produkuje również metryki jakości, w tym korelację Spearmana i głębokość odczytu. | |||
Dotyczące wyrównania i kwantyfikacji:
Mapowanie odczytów odbywa się za pomocą programu STAR (w niektórych przypadkach używane są zarówno alignery STAR, jak i TopHat, które tworzą oddzielne pliki bam), a kwantyfikacja genów i transkryptów odbywa się za pomocą programu RSEM. Chociaż istnieje ogólna zgodność pomiędzy mapowaniami i kwantyfikacjami genów, tworzonymi przez różne potoki RNA-seq, kwantyfikacje poszczególnych izoform transkryptów, jako znacznie bardziej złożone, mogą się znacznie różnić w zależności od zastosowanego potoku przetwarzania i ich dokładność jest nieznana. Dlatego też, alignacje i kwantyfikacje genów mogą być używane pewnie, podczas gdy kwantyfikacje transkryptów powinny być używane ostrożnie.
Odniesienia genomowe
Zobacz odniesienia do genomów i rozmiary chromosomów używane w tym potoku
Potoki te wymagają zarówno informacji o montażu dla interesującego nas gatunku, jak i odniesienia do genu. Każdy z głównych programów, TopHat, STAR i RSEM, tworzy indeks do wykorzystania w kolejnych krokach. Więcej informacji na temat użycia RSEM jest dostępnych tutaj.
Ekogeniczne kontrole RNA typu spike-in
Ekogeniczne kontrole RNA typu spike-in są dodawane do próbek w celu stworzenia standardowej linii bazowej dla kwantyfikacji ekspresji RNA (PMC3166838). Konsorcjum ENCODE standaryzuje się na użycie dostępnych na rynku spike-inów Ambion Mix 1 w rozcieńczeniu ~2% końcowych zmapowanych odczytów. Jednakże, istnieje mieszanka starszych danych i danych importowanych. Dlatego, aby śledzić spike-iny użyte w danej bibliotece, istnieje zbiór danych powiązany z tą biblioteką. Ten zbiór danych będzie zawierał plik sekwencji spike-ins w formacie fasta oraz informacje o stężeniach. Oczekuje się, że te sekwencje spike-in znajdą się w indeksie genomu użytym w kroku(ach) mapowania i w wygenerowanych następnie bam. Kwantyfikacje sekwencji można znaleźć w plikach kwantyfikacji transkryptów i genów RSEM.
Zobacz zestawy danych spike-in
Zobacz certyfikat analizy dla ERCC spike-in
Dostęp do ERCC dash board
Linki i publikacje
Znajdź dane wygenerowane przez ten potok: All | paired-end only | single-end only
Eksploruj publikacje (w toku)
.