
Vizualizare generală a conductei
Conducta de procesare a datelor de ARN-seq în masă a fost dezvoltată ca parte a seriei ENCODE Uniform Processing Pipelines. Codul complet al conductei este disponibil în mod gratuit pe Github și poate fi rulat pe DNAnexus (linkul necesită crearea unui cont) la prețul lor actual.
The ENCODE Bulk RNA-seq pipeline poate fi utilizat atât pentru bibliotecile de ARN-seq replicat și nereplicat, împerecheate sau cu un singur capăt, precum și pentru bibliotecile de ARN-seq specifice sau nespecifice de catenă. Bibliotecile trebuie să fie generate din ARNm (ARN poli(A)+, ARN total sărăcit de ARNr sau populații de ARN poli(A) care sunt selectate în funcție de dimensiune pentru a fi mai lungi de aproximativ 200 bp. În viitor, această conductă poate fi utilizată, de asemenea, pentru a procesa datele PAS-seq și Bru-seq.
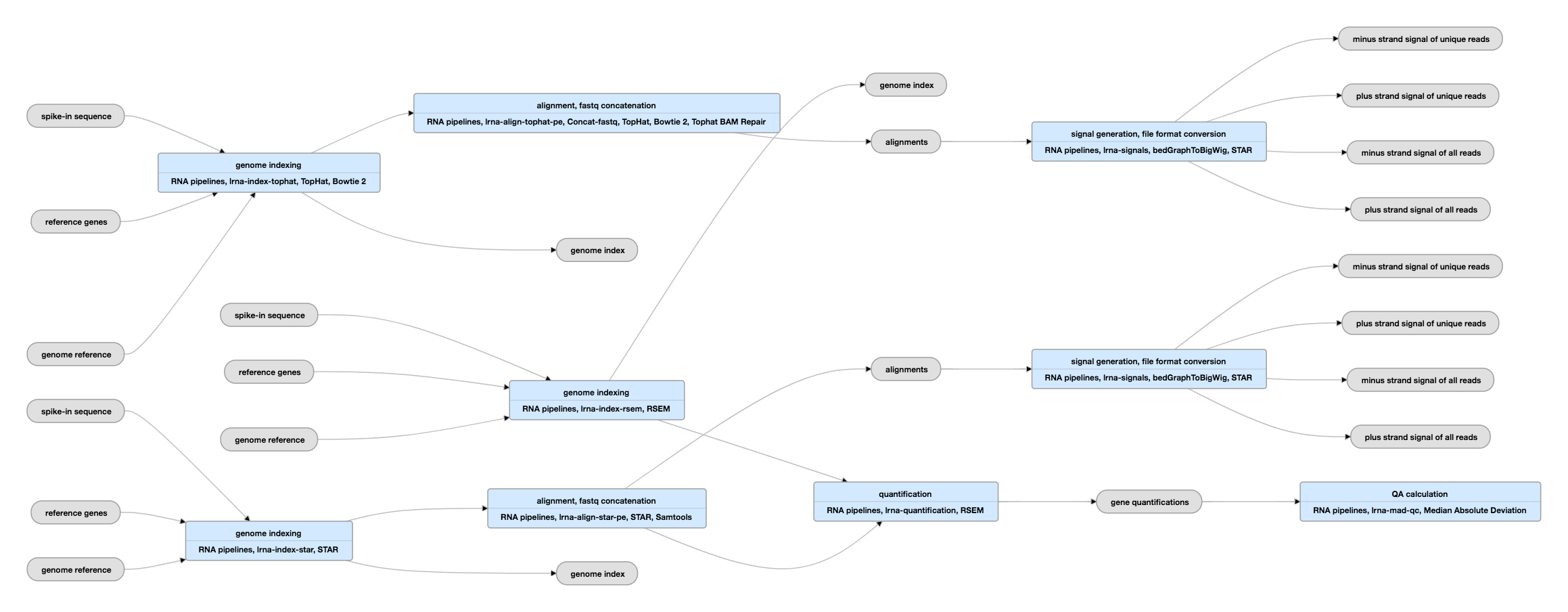
Pipeline Schematic for paired-ended data
Vezi instanța actuală a acestei conducte pentru date împerecheate
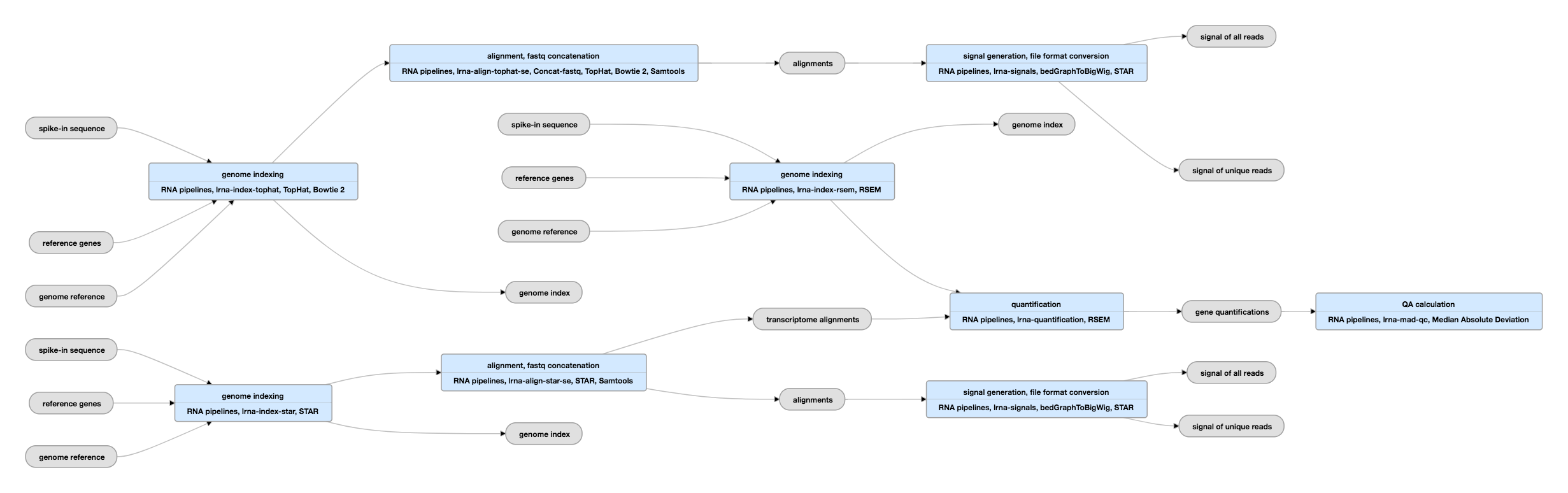
Pipeline Schematic for single-ended data
Vezi instanțele actuale ale acestei conducte pentru date cu un singur capăt
Intrări:
| Fișier format |
Informații conținute în fișier |
Fișier description |
Note |
| fastq |
reads |
G-zipped bulk RNA-seq reads | Lecturile trebuie să îndeplinească criteriile descrise în Uniform Processing Pipeline Restrictions. |
| tar | genome index | Generat de STAR sau TopHat | Vă rugăm să consultați paragraful intitulat „Regarding alignment and quantification” (În ceea ce privește alinierea și cuantificarea) de sub tabelul „Outputs” (Rezultate) pentru mai multe informații despre aliniere și indicii acestora. |
| fasta | speke-in sequence | ERCC Spike-ins (External RNA Control Consortium) | Speke-ins sunt efectiv controalele pentru experimentul RNA-seq. |
Outputs:
| File format |
Informații conținute în fișier |
Descriere fișier |
Note |
| bam | aliniamente | Produse prin cartografierea citirilor la genom. | Vă rugăm să consultați paragraful intitulat „Regarding alignment and quantification” (În ceea ce privește alinierea și cuantificarea) de sub tabelul „Outputs” (Rezultate) pentru mai multe informații despre aliniatori și indicii acestora. |
| bam | aliniamente de transcriptom | Produse prin cartografierea citirilor la transcriptom. | |
| bigWig | signal | Semnalul RNA-seq normalizat | Pentru datele filamentare, se generează semnale pentru citirile unice și citirile unice+multimaptare atât în filamentul pozitiv, cât și în cel negativ. Pentru datele necatenare, se generează semnale pentru citirile unice și citirile unice+multimapping fără a se ține cont de identitatea catenei. |
| tsv | cantificări de gene | Include cuantificările spike-ins |
Specificațiile formatului de fișier sunt următoarele:
|
| tsv | cantificări de transcripte | Include cuantificările spike-ins | Vă rugăm să consultați avertismentul privind cuantificările de transcripte din paragraful de mai jos intitulat „Cu privire la aliniere și cuantificare”. |
| Peline-ul produce, de asemenea, măsurători de calitate, inclusiv corelația Spearman și adâncimea de citire. | |||
În ceea ce privește alinierea și cuantificarea:
Corectarea citirilor se face cu ajutorul programului STAR (în unele cazuri, se folosesc atât STAR cât și alinierii TopHat pentru a produce fișiere bam separate), iar cuantificarea genelor și a transcriptelor se face cu programul RSEM. Deși există o concordanță generală între cartografieri și cuantificările genelor produse de diferite pipeline-uri de ARN-seq, cuantificările izoformelor individuale ale transcriptelor, fiind mult mai complexe, pot diferi substanțial în funcție de pipeline-ul de procesare utilizat și au o precizie necunoscută. Prin urmare, alinierile și cuantificările de gene pot fi utilizate cu încredere, în timp ce cuantificările de transcripte ar trebui utilizate cu grijă.
Genomic References
Vezi referințele genomului și dimensiunile cromozomilor utilizate în această conductă
Aceste conducte necesită atât informații de asamblare pentru specia de interes, cât și o referință genetică. Fiecare dintre programele principale, TopHat, STAR și RSEM creează un index pentru a fi utilizat în etapele următoare. Mai multe informații cu privire la utilizarea RSEM sunt disponibile aici.
Controale de spike-in cu ARN exogen
Controale de spike-in cu ARN exogen sunt adăugate la probe pentru a crea o linie de bază standard pentru cuantificarea expresiei ARN (PMC3166838). Consorțiul ENCODE standardizează utilizarea amestecului Ambion Mix 1, disponibil în comerț, la o diluție de ~2% din citirile cartografiate finale. Cu toate acestea, există un amestec de date mai vechi și date importate. Prin urmare, pentru a urmări spike-in-urile utilizate într-o anumită bibliotecă, există un set de date asociat cu biblioteca respectivă. Setul de date respectiv va conține fișierul cu secvența de spike-ins în format fasta și informații privind concentrațiile. Se așteaptă ca aceste secvențe spike-in să se regăsească în indexul genomului utilizat în etapa (etapele) de cartografiere și în bam-ul generat ulterior. Cuantificările secvențelor pot fi găsite în fișierele RSEM de cuantificare a transcriptelor și a genelor.
Vezi seturile de date spike-ins
Vezi certificatul de analiză pentru ERCC spike-ins
Accesați tabloul de bord ERCC
Links and Publications
Căutați datele generate de acest pipeline: All | paired-end only | single-end only
Explorați publicațiile (în curs)
.