
Översikt över pipeline
Pipeline för Bulk RNA-seq utvecklades som en del av ENCODE:s serie Uniform Processing Pipelines. Den fullständiga pipelinekoden är fritt tillgänglig på Github och kan köras på DNAnexus (länken kräver att ett konto skapas) till deras nuvarande pris.
EnCODE Bulk RNA-seq pipeline kan användas för både replikerade och icke replikerade, parvisa eller enkelriktade och strängspecifika eller icke strängspecifika RNA-seq-bibliotek. Biblioteken måste genereras från mRNA (poly(A)+, rRNA-depleted total RNA eller poly(A)-populationer som är storleksselekterade så att de är längre än cirka 200 bp. I framtiden kan denna pipeline också användas för att bearbeta PAS-seq- och Bru-seq-data.
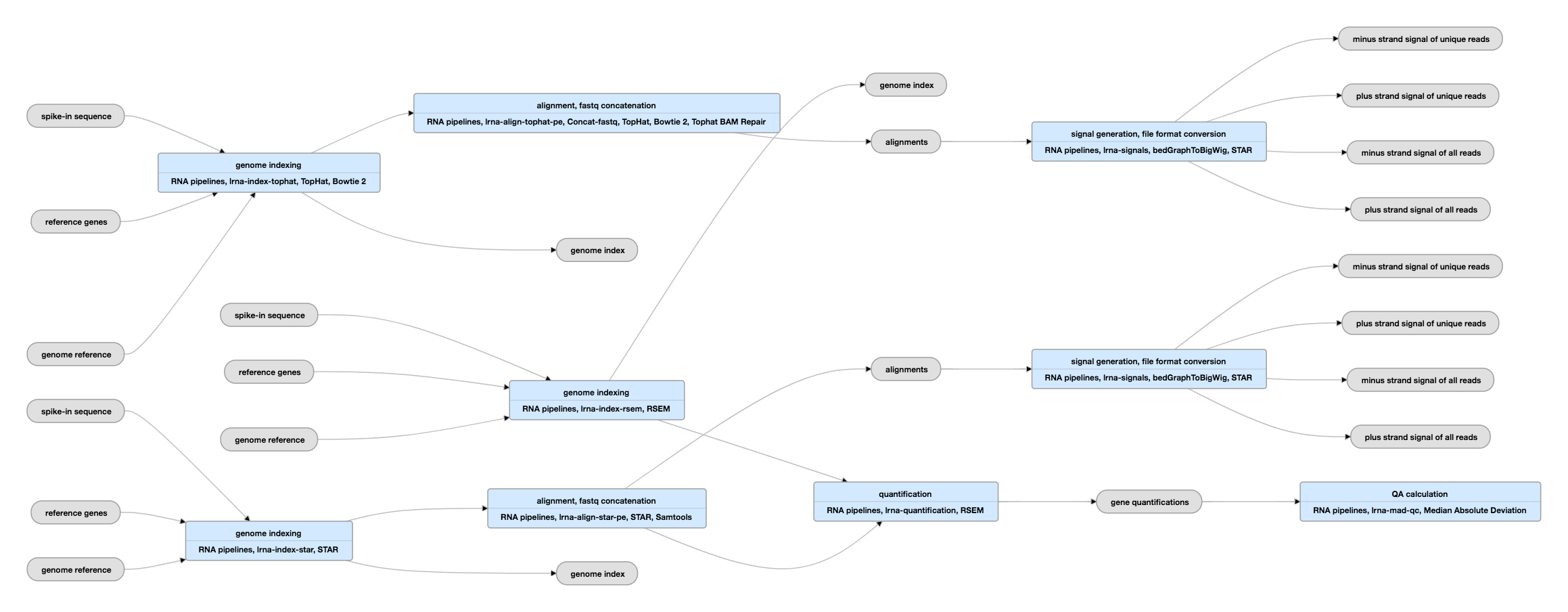
Schematisk pipeline för data med parade ändar
Se den aktuella instansen av denna pipeline för data med parade ändar
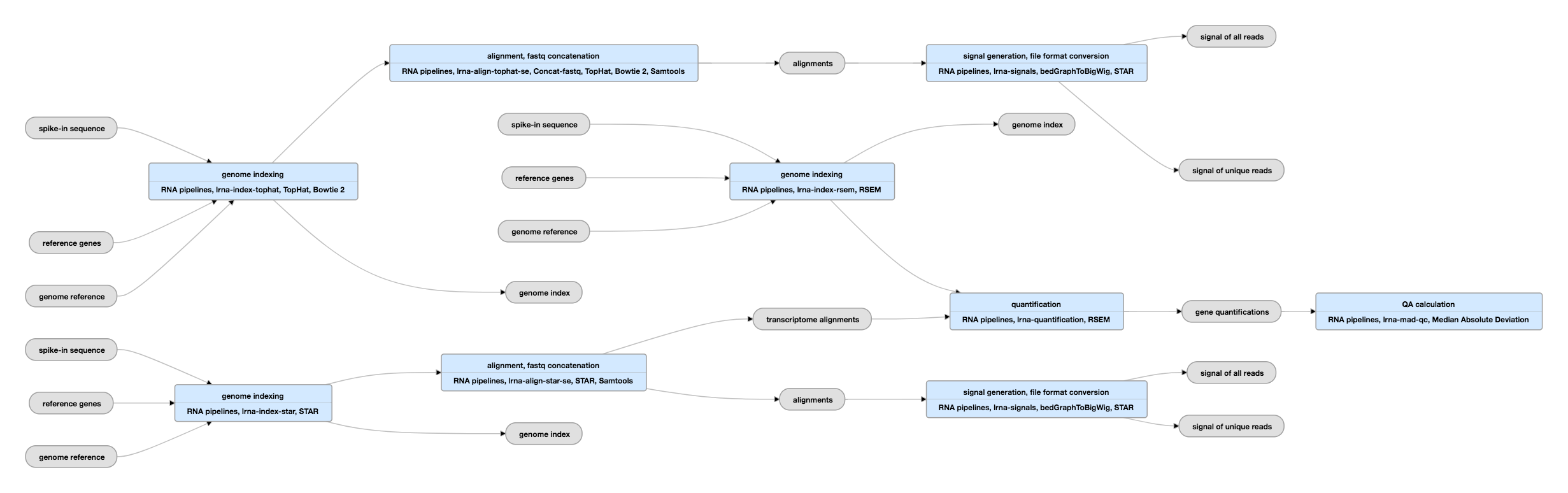
Skematisk pipeline för data med enkla ändar
Se de aktuella instanserna av denna pipeline för data med enkla ändar
Inputs:
| Filformat |
Information i filen |
Fil Beskrivning |
Anteckningar |
| fastq |
läser |
G-Zippade RNA-seq-avläsningar i bulk | Avläsningarna måste uppfylla de kriterier som anges i Uniform Processing Pipeline Restrictions. |
| tar | genomindex | Genererat av STAR eller TopHat | Se stycket med rubriken ”Regarding alignment and quantification” (Angående anpassning och kvantifiering) under tabellen ”Outputs” (utdata) för mer information om aligners och deras index. |
| fasta | spike-in-sekvens | ERCC Spike-ins (External RNA Control Consortium) | Spike-ins är i praktiken kontroller för RNA-seq-experimentet. |
Outputs:
| Filformat |
Information i filen |
Filbeskrivning |
Anteckningar |
| bam | utjämning | Producerad genom att mappa läsningar till genomet. | Se stycket med rubriken ”Angående anpassning och kvantifiering” under tabellen ”Resultat” för mer information om anpassarna och deras index. |
| bam | transkriptomanpassningar | Framställs genom att mappa avläsningar till transkriptomet. | |
| bigWig | signal | Normaliserad RNA-seq-signal | För strängdata genereras signaler för unika läsningar och unika+multimappningsläsningar i både plus- och minussträngen. För osträngade data genereras signaler för unika läsningar och unika+multimapping läsningar utan hänsyn till strängidentitet. |
| tsv | genkvantifieringar | Inkluderar spike-ins-kvantifieringar |
Specifikationerna för filformatet är följande:
|
| tsv | transkriptkvantifieringar | Inkluderar spike-ins-kvantifieringar | Vänligen se försiktighet angående transkriptkvantifieringar i stycket nedan med titeln ”Regarding alignment and quantification”. |
| Pipeline producerar också kvalitetsmått, inklusive Spearmankorrelation och läsdjup. | |||
Regering av anpassning och kvantifiering:
Kartläggningen av läsningarna görs med hjälp av STAR-programmet (i vissa fall används både STAR- och TopHat-anpassare för att producera separata bam-filer) och kvantifieringen av gener och transkriptioner görs med RSEM-programmet. Även om det finns en allmän överensstämmelse mellan de kartläggningar och genkvantifieringar som produceras av olika RNA-seq-pipelines, kan kvantifieringarna av enskilda transkriptisoformer, som är mycket mer komplexa, skilja sig avsevärt beroende på vilken bearbetnings pipeline som används och har en okänd noggrannhet. Därför kan anpassningar och genkvantifieringar användas med säkerhet, medan transkriptkvantifieringar bör användas med försiktighet.
Genomreferenser
Se de genomreferenser och kromosomstorlekar som används i denna pipeline
Dessa pipelines kräver både information om sammansättning för den aktuella arten och en genreferens. Var och en av huvudprogrammen TopHat, STAR och RSEM skapar ett index som används i efterföljande steg. Mer information om användningen av RSEM finns här.
Exogen RNA spike-in-kontroller
Exogen RNA spike-in-kontroller läggs till prover för att skapa en standardbaslinje för kvantifiering av RNA-uttryck (PMC3166838). ENCODE-konsortiet standardiserar användningen av Ambion Mix 1 kommersiellt tillgängliga spike-ins i en utspädning på ~2 % av de slutliga kartlagda läsningarna. Det finns dock en blandning av äldre data och importerade data. För att spåra de spike-ins som används i ett visst bibliotek finns det därför ett dataset som är kopplat till biblioteket. Datasetetet kommer att innehålla sekvensfilen för spike-ins i fasta-format och information om koncentrationerna. Dessa spike-in-sekvenser förväntas återfinnas i det genomindex som används i kartläggningssteget eller -stegen och i den därefter genererade bam-filen. Kvantifieringarna av sekvenserna finns i RSEM:s transkript- och genkvantifieringsfiler.
Se spike-ins-dataset
Se analyscertifikatet för ERCC spike-ins
Access the ERCC dash board
Länkar och publikationer
Sök efter data som genererats av den här pipelinen: Alla | endast parade ändar | endast enändiga ändar
Utforska publikationer (pågående)