
- Genom Sequencing
- Genetic Characterization
- Methods of Flu Genome Sequencing
Genome Sequencing
Influensavirus förändras ständigt, I själva verket genomgår alla influensavirus genetiska förändringar med tiden (för mer information, se Hur influensaviruset kan förändras: ”Drift” och ”Shift”). Ett influensavirus genom består av alla gener som ingår i viruset. CDC bedriver året runt övervakning av cirkulerande influensavirus för att övervaka förändringar i genomet (eller delar av genomet) hos dessa virus. Detta arbete utförs som en del av den rutinmässiga amerikanska influensaövervakningen och som en del av CDC:s roll som ett av Världshälsoorganisationens (WHO) Collaborating Center for Reference and Research on Influenza (samarbetscentrum för referens och forskning om influensa). Den information som CDC samlar in genom att studera genetiska förändringar (även kallade ”substitutioner”, ”varianter” eller ”mutationer”) i influensavirus spelar en viktig roll för folkhälsan genom att hjälpa till att avgöra om vacciner och antivirala läkemedel kommer att fungera mot för närvarande cirkulerande influensavirus, samt hjälpa till att fastställa potentialen för influensavirus hos djur att infektera människor.
Genomsekvensering avslöjar sekvensen av nukleotider i en gen, som alfabetbokstäver i ord. Nukleotider är organiska molekyler som utgör den strukturella enhetsbyggstenen i nukleinsyror, t.ex. RNA eller DNA. Alla influensavirus består av enkelsträngat RNA i motsats till dubbelsträngat DNA. Influensavirusens RNA-gener består av kedjor av nukleotider som är bundna till varandra och kodas av bokstäverna A, C, G och U, som står för adenin, cytosin, guanin respektive uracil. Genom att jämföra sammansättningen av nukleotider i en virusgen med ordningen av nukleotider i en annan virusgen kan man avslöja variationer mellan de två virusen.
Genetiska variationer är viktiga eftersom de kan påverka strukturen hos ett influensavirus ytproteiner. Proteiner består av sekvenser av aminosyror.
Substitutionen av en aminosyra mot en annan kan påverka egenskaper hos ett virus, t.ex. hur väl ett virus överförs mellan människor och hur mottagligt viruset är för antivirala läkemedel eller aktuella vacciner.
![]()
![]()
Genomsekvensering avslöjar sekvensen av nukleotiderna i en gen, som alfabetbokstäver i ord. Genom att jämföra sammansättningen av nukleotider i en virusgen med ordningen av nukleotider i en annan virusgen kan man avslöja variationer mellan de två virusen.
Geniska variationer är viktiga eftersom de påverkar strukturen av ett influensavirus ytproteiner. Proteiner består av sekvenser av aminosyror.
Substitutionen av en aminosyra mot en annan kan påverka egenskaper hos ett virus, t.ex. hur väl ett virus överförs mellan människor och hur mottagligt viruset är för antivirala läkemedel eller aktuella vacciner.
Influensa A- och B-virus – de primära influensavirusen som smittar människor – är RNA-virus som har åtta gensegment. Dessa gener innehåller ”instruktioner” för att skapa nya virus, och det är dessa instruktioner som ett influensavirus använder när det infekterar en mänsklig cell för att lura cellen att producera fler influensavirus och på så sätt sprida infektionen.
Influensagenerna består av en sekvens av molekyler som kallas nukleotider och som binds samman i en kedjeliknande form. Nukleotider betecknas med bokstäverna A, C, G och U.
Genomsekvensering är en process som bestämmer ordningen, eller sekvensen, av nukleotiderna (dvs. A, C, G och U) i var och en av de gener som finns i virusets genom. Fullgenomsekvensering kan avslöja den cirka 13 500 bokstäver långa sekvensen av alla gener i virusets genom.
Varje år utför CDC helgenomsekvensering av cirka 7 000 influensavirus från ursprungliga kliniska prover som samlats in genom virologisk övervakning. Ett influensa A- eller B-virus’ genom innehåller åtta gensegment som kodar för (dvs. bestämmer strukturen och egenskaperna hos) virusets 12 proteiner, inklusive dess två primära ytproteiner: hemagglutinin (HA) och neuraminidas (NA). Ett influensavirus ytproteiner bestämmer viktiga egenskaper hos viruset, bland annat hur viruset reagerar på vissa antivirala läkemedel, virusets genetiska likhet med nuvarande influensavaccinvirus och potentialen för zoonotiska (av djur härstammande) influensavirus att infektera mänskliga värdar.
Genetisk karakterisering
CDC och andra folkhälsolaboratorier runt om i världen har sekvenserat generna hos influensavirus sedan 1980-talet. CDC bidrar med gensekvenser till offentliga databaser, t.ex. GenBankextern ikon och Global Initiative on Sharing Avian Influenza Data (GISAID)extern ikon, för användning av folkhälsoforskare. De resulterande biblioteken med gensekvenser gör det möjligt för CDC och andra laboratorier att jämföra generna hos de influensavirus som för närvarande cirkulerar med generna hos äldre influensavirus och virus som används i vacciner. Denna process för att jämföra genetiska sekvenser kallas genetisk karakterisering. CDC använder sig av genetisk karakterisering av följande skäl:
- För att fastställa hur nära ”besläktade” eller liknande influensavirus är varandra genetiskt
- För att övervaka hur influensavirus utvecklas
- För att identifiera genetiska förändringar som påverkar virusets egenskaper. Till exempel för att identifiera de specifika förändringar som är förknippade med influensavirus som sprids lättare, orsakar allvarligare sjukdom eller utvecklar resistens mot antivirala läkemedel
- För att bedöma hur väl ett influensavaccin mot influensa skulle kunna skydda mot ett visst influensavirus baserat på dess genetiska likhet med viruset
- För att övervaka om det finns genetiska förändringar i influensavirus som cirkulerar i djurpopulationer som skulle kunna göra det möjligt för dem att infektera människor.
De relativa skillnaderna mellan en grupp influensavirus visas genom att organisera dem i ett diagram som kallas ”fylogenetiskt träd”. Fylogenetiska träd för influensavirus är som släktträd (genealogi) för människor. Träden visar hur nära ”besläktade” enskilda virus är med varandra. Virusen grupperas tillsammans baserat på om deras geners nukleotider är identiska eller inte. Fylogenetiska träd för influensavirus visar vanligtvis hur lika virusens hemagglutinin- (HA) eller neuraminidasgener (NA) är varandra. Varje sekvens från ett specifikt influensavirus har en egen gren på trädet. Graden av genetisk skillnad (antalet nukleotidskillnader) mellan virus representeras av längden på de horisontella linjerna (grenarna) i det fylogenetiska trädet. Ju längre virusen befinner sig från varandra på den horisontella axeln i ett fylogenetiskt träd, desto mer genetiskt olika är virusen från varandra.
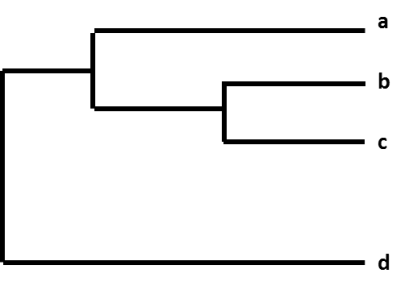
Figur. Ett fylogenetiskt träd.
När CDC har sekvenserat ett influensa A(H3N2)-virus som samlats in genom övervakning katalogiseras virussekvensen tillsammans med andra virussekvenser som har en liknande HA-gen (H3) och en liknande NA-gen (N2). Som en del av denna process jämför CDC den nya virussekvensen med de andra virussekvenserna och letar efter skillnader mellan dem. CDC använder sedan ett fylogenetiskt träd för att visuellt representera hur genetiskt olika A(H3N2)-virus är från varandra.
CDC utför genetisk karakterisering av influensavirus året runt. Dessa genetiska uppgifter används tillsammans med uppgifter om virusens antigena karakterisering för att hjälpa till att avgöra vilka vaccinvirus som ska väljas för de kommande influensavaccinerna för norra respektive södra halvklotet. Under månaderna före WHO:s samrådsmöten om vaccin i februari och september samlar CDC in influensavirus genom övervakning och jämför HA- och NA-gensekvenserna hos aktuella vaccinvirus med sekvenserna hos cirkulerande influensavirus. Detta är ett sätt att bedöma hur nära besläktade de cirkulerande influensavirusen är med de virus som säsongsinfluensavaccinet utformades för att skydda mot. När virus samlas in och karakteriseras genetiskt kan skillnader avslöjas.
Till exempel förändras cirkulerande virus ibland under loppet av en säsong genetiskt, vilket gör att de skiljer sig från motsvarande vaccinvirus. Detta är en indikation på att ett annat vaccinvirus kan behöva väljas ut för nästa influensasäsongs vaccin, även om andra faktorer, inklusive resultat av antigena karakteriseringar, i hög grad påverkar vaccinbesluten. HA- och NA-ytproteinerna i influensavirus är antigener, vilket innebär att de känns igen av immunsystemet och kan utlösa ett immunsvar, inklusive produktion av antikroppar som kan blockera infektionen. Antigenisk karakterisering avser analysen av ett virus reaktion med antikroppar för att hjälpa till att bedöma hur det förhåller sig till ett annat virus.
Metoder för sekvensering av influensagenomen
Ett influensaprov innehåller många influensaviruspartiklar som odlats i ett provrör och som ofta har små genetiska skillnader i jämförelse med varandra bland hela populationen av syskonvirus.
Traditionsenligt har forskare använt en sekvenseringsteknik som kallas ”Sangerreaktionen” för att övervaka influensans utveckling som en del av den virologiska övervakningen. Sangersekvensering identifierar den dominerande genetiska sekvensen bland de många influensavirus som finns i ett isolat. Detta innebär att små variationer i populationen av virus som finns i ett prov inte återspeglas i slutresultatet. Forskare använder ofta Sanger-metoden för att utföra partiell genomsekvensering av influensavirus, medan nyare teknik (se nästa stycke) är bättre lämpad för sekvensering av hela genomet.
Under de senaste fem åren har CDC använt sig av ”Next Generation Sequencing (NGS)”-metoder, som kraftigt har utökat mängden information och detaljer som sekvenseringsanalyser kan ge. NGS använder avancerad molekylär detektion (AMD) för att identifiera gensekvenser från varje virus i ett prov. Därför avslöjar NGS de genetiska variationerna bland många olika influensaviruspartiklar i ett enda prov, och dessa metoder avslöjar också hela den kodande regionen i genomerna. Denna detaljnivå kan direkt gynna beslutsfattandet inom folkhälsan på viktiga sätt, men data måste tolkas noggrant av högt utbildade experter i samband med annan tillgänglig information. Se AMD-projekt: Förbättra influensavaccin för mer information om hur NGS och AMD revolutionerar kartläggningen av influensagenomen vid CDC.