
Pipeline overview
O gasoduto Bulk RNA-seq foi desenvolvido como parte da série ENCODE Uniform Processing Pipelines. O código completo do pipeline está disponível gratuitamente no Github e pode ser executado no DNAnexus (o link requer a criação de conta) ao seu preço atual.
O gasoduto ENCODE bulk RNA-seq pode ser usado tanto para bibliotecas de RNA-seq replicadas como não replicadas, com ou sem ponta, e bibliotecas de RNA-seq específicas ou sem ponta. As bibliotecas devem ser geradas a partir de mRNA (poli(A)+, rRNA total descarregado, ou poli(A)- populações de tamanho selecionado para ser maior que aproximadamente 200 bp. No futuro, esse pipeline também poderá ser usado para processar dados PAS-seq e Bru-seq.
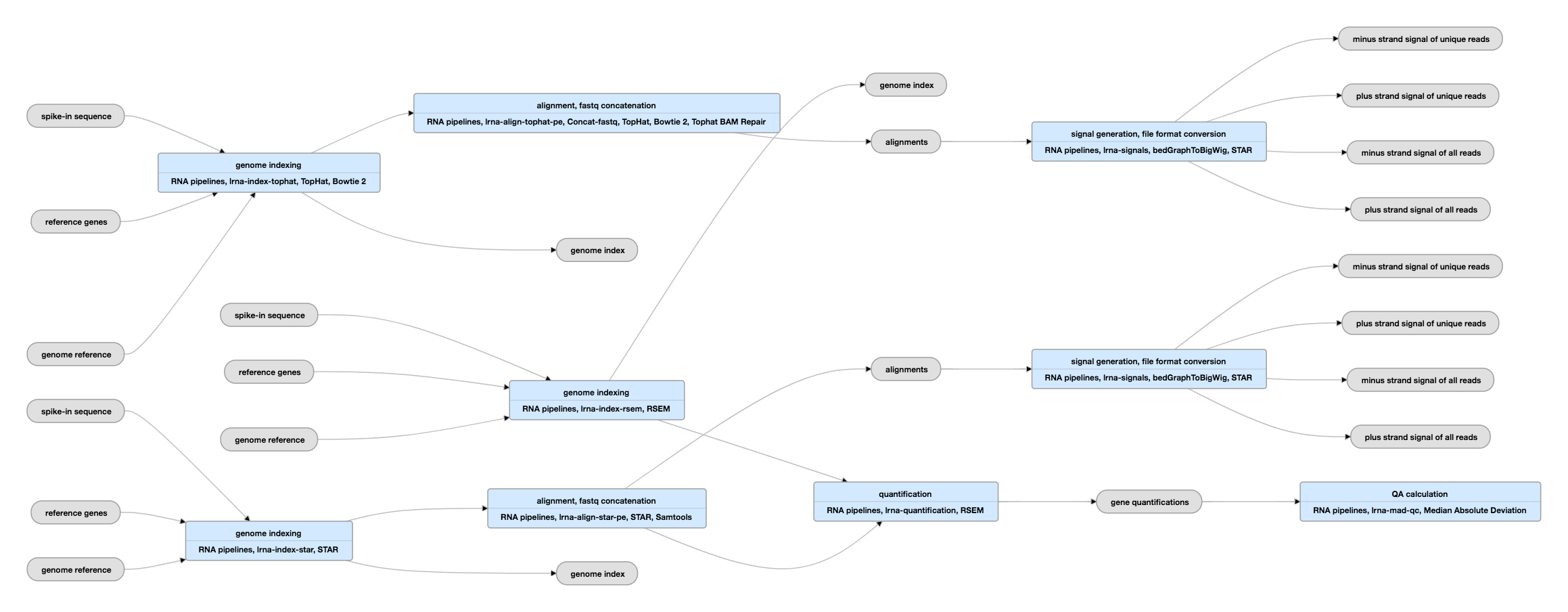
Esquema-pipeline Esquema para dados com ponta de pizza
Veja a instância atual deste pipeline para dados com ponta de pizza
 >
>
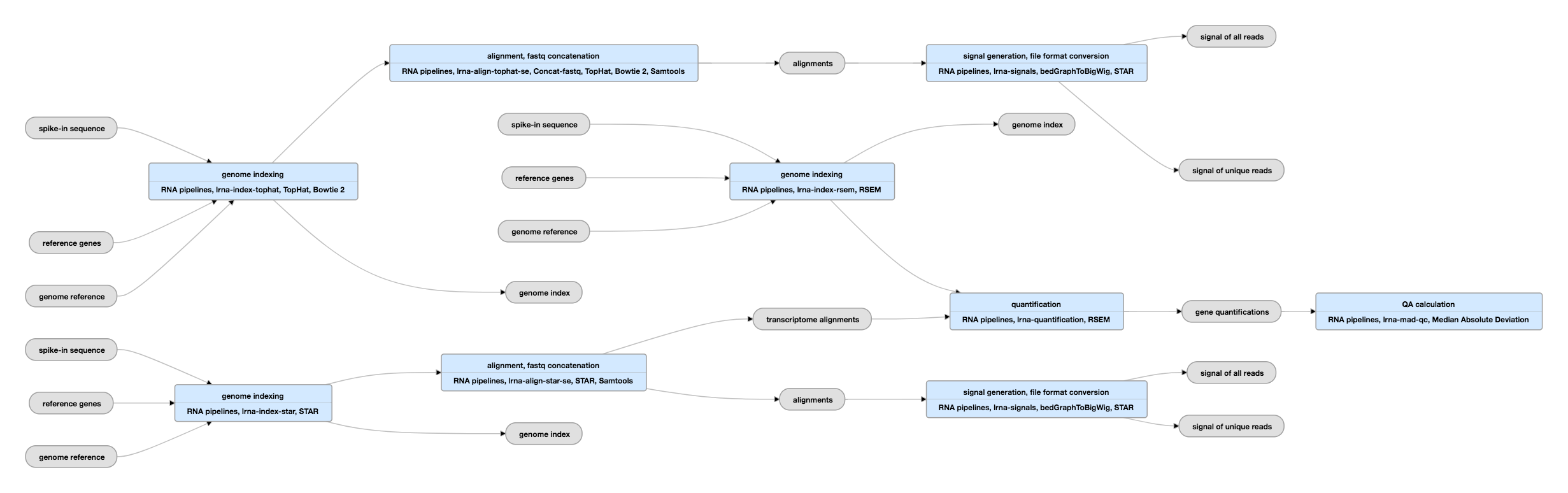
Esquema-pipeline para dados com ponta única
Veja as instâncias atuais deste pipeline para dados com ponta única
Entradas:
| Formato de arquivo |
Informação contida no arquivo |
Arquivo descrição |
Notas |
| fastq |
reads |
G-O RNA-seq de volume compactado lê | As leituras devem atender aos critérios descritos nas Restrições de Processamento Uniforme de Dutos. |
| tar | Índice de genoma | Gerado por STAR ou TopHat | Por favor veja o parágrafo intitulado “Sobre alinhamento e quantificação” abaixo da tabela “Outputs” para mais informações sobre os alinhadores e seus índices. |
| fasta | seqüencia de Spike-in | Spike-ins ERCC (External RNA Control Consortium) | Os spike-ins são efetivamente os controles para o experimento RNA-seq. |
Saídas:
| Formato do arquivo |
Informação contida no arquivo |
Descrição do arquivo |
Notas |
| bam | alinhamentos | Produzido pelo mapeamento de leituras para o genoma. | Por favor veja o parágrafo intitulado “Sobre alinhamento e quantificação” abaixo da tabela “Outputs” para mais sobre os alinhadores e seus índices. |
| bam | alinhamentos do transriptoma | Produzido pelo mapeamento de leituras para o transriptoma. | |
| bigWig | sinal | Sinal RNA-seq normalizado | Para dados encalhados, são gerados sinais para leituras únicas e leituras únicas+multimapping em ambas as vertentes mais e menos. Para dados não encalhados, os sinais são gerados para leituras únicas e leituras únicas+multimapping sem considerar a identidade do encadeamento. |
| tsv | quantificações genéticas | Inclui as quantificações de picos |
As especificações do formato do ficheiro são as seguintes:
>coluna 7: FPKM (fragmentos por quilobase de transcrição por milhão)>coluna 8: contagem_de_sentido_posterior >coluna 11: pme_FPKM |
| tsv | quantificações transcritas | Inclui as quantificações de picos | Por favor veja o cuidado com as quantificações transcritas no parágrafo abaixo intitulado “Sobre alinhamento e quantificação”. |
| O gasoduto também produz métricas de qualidade, incluindo a correlação Spearman e a profundidade de leitura. | |||
Reglar alinhamento e quantificação:
O mapeamento das leituras é feito usando o programa STAR (em alguns casos, ambos alinhadores STAR e TopHat são usados para produzir arquivos bam separados) e a quantificação dos genes e transcrições é feita com o programa RSEM. Embora exista uma concordância geral entre os mapeamentos e as quantificações de genes produzidos por diferentes dutos RNA-seq, as quantificações de isoformas de transcrição individuais, sendo muito mais complexas, podem diferir substancialmente dependendo do dutos de processamento empregado e são de precisão desconhecida. Portanto, alinhamentos e quantificações de genes podem ser usados com confiança, enquanto que as quantificações de transcrições devem ser usadas com cuidado.
Referências Genômicas
Ver as referências genômicas e tamanhos de cromossomos usados neste pipeline
Estes pipelines requerem tanto informações de montagem para as espécies de interesse como uma referência gênica. Cada um dos programas principais, TopHat, STAR, e RSEM criam um índice para uso em etapas subsequentes. Mais informações sobre o uso do RSEM estão disponíveis aqui.
Controles spike-in de RNA exógeno
Controles spike-in de RNA exógeno são adicionados às amostras para criar uma linha de base padrão para a quantificação da expressão do RNA (PMC3166838). O consórcio ENCODE está a padronizar a utilização do Ambion Mix 1, com uma diluição de ~2% das leituras finais mapeadas. No entanto, existe uma mistura de dados mais antigos e dados importados. Portanto, para rastrear os spike-ins usados em uma determinada biblioteca, há um conjunto de dados associados à biblioteca. Esse conjunto de dados conterá o arquivo de seqüência de spike-ins em formato fasta e informações sobre as concentrações. Estas sequências de spike-ins devem ser encontradas no índice do genoma utilizado na(s) etapa(s) do mapeamento e no bam gerado posteriormente. As quantificações das sequências podem ser encontradas nos arquivos de transcrição RSEM e quantificação de genes.
Visualizar conjuntos de dados de spike-ins
Visualizar o certificado de análise para spike-ins ERCC
Acesso ao painel de controle ERCC
Links e Publicações
Visualizar dados gerados por este pipeline: Todos |Apenas ponta-a-ponta |Apenas ponta-a-ponta
Explorar publicações (em progresso)